Regular Articles
Virtual Screening with Docking Simulations and Biochemical Evaluation of VHY Phosphatase Inhibitors
2015 Volume 63 Issue 10 Pages 807-811
Details
2015 Volume 63 Issue 10 Pages 807-811
Although VH1-related member Y (VHY) phosphatase is responsible for the pathogenesis of neuroinflammatory diseases, no small-molecule inhibitor of VHY has been reported so far. Here we first report eight VHY inhibitors identified from molecular docking-based virtual screening and subsequent enzyme inhibition assays. These inhibitors exhibit good biochemical potencies against VHY, with associated IC50 values ranging from 1 to 9 µM. Because all these inhibitors were also screened in silico for having desirable physicochemical properties as a drug candidate, they deserve further investigation by structure–activity relationship studies to develop new medicines for the treatment of neuroinflammatory diseases. The structural features of VHY-inhibitor interactions relevant to the micromolar-level inhibitory activity are addressed in detail.
Dual-specificity protein phosphatases (DUSPs) catalyze the hydrolysis reactions of the phosphorylated serine/threonine and tyrosine residues in protein substrates, which serves as a hallmark of various cellular signal transductions.1) Atypical DUSPs are structurally different from typical ones in that they have a single catalytic domain only and lack the kinase-binding domain required for specific binding of substrates.2) VH1-related member Y (VHY), which is also known as DUSP15, belongs to the atypical DUSP group and is an active phosphatase readily detectable in spermatocytes and spermatids.3)
Besides the involvement in regulating the meiotic signal transduction in testis cells, it was found in the systematic genomic analyses to investigate the activity of PTP genes that VHY would also be a key regulator of oligodendrocyte differentiation.4) Because oligodendrocyte mediates the remyelination in the nerve system with myelin damage, VHY has been considered as a promising therapeutic target protein for the treatment of multiple sclerosis, which is a neuroinflammatory disease characterized by the progressive loss of myelin.
Three dimensional structure of human VHY was reported in complex with the sulfate ion that had been used as an effective surrogate for the phosphate group in the substrates.5) Overall structure of VHY appeared to be similar to that of VH1-related dual-specific protein phosphatase (VHR)6): root-mean-square deviation between Cα atoms of the two DUSPs amounted to only 1.6 Å. Nonetheless, amino acid sequences of the two atypical DUSPs are quite different around the active site including the phosphatase (P) loop. The P loop of VHY (Phe89–Ala90–Gly91–Ile92–Ser93) contains more hydrophobic residues than that of VHR (Arg125–Glu126–Gly127–Tyr128–Gly129). Such detailed structural information on the active site structure and the interaction with a small-molecule ligand can be useful for designing the potent VHY inhibitors that may develop into a drug candidate for the treatment of multiple sclerosis. However, the discovery of VHY inhibitors lags behind the biological and structural studies. As a matter of fact, no small-molecule VHY inhibitor has been reported so far neither in the literature nor in patents.
In this study, we aim to identify the novel classes of VHY inhibitors be means of virtual screening with molecular docking and biochemical evaluation of the computationally screened molecules. Molecular docking-based virtual screening has not always been successful due to the imperfections in the scoring function to estimate the binding affinity between the target protein and a ligand. This leads to a poor correlation between the computational and experimental results for binding affinities.7) Recently we developed a modified AutoDock scoring function by replacing the existing solvation free energy term with the more accurate one developed based on the extended solvent-contact model.8) This new scoring function has proved to outperform the previous one in predicting the binding affinities of small-molecule ligands with respect to various target proteins.9,10) It will be shown in this study that the molecular docking with the modified scoring function can be a useful virtual screening tool for enriching the chemical library with molecules that have good inhibitory activity against VHY as well as for elucidating the binding affinities of the identified inhibitors.
X-Ray crystal structure of human VHY in complex with the sulfate ion (PDB code: 1YZ4)4) was used as the receptor model in molecular docking for virtual screening of VHY inhibitors. The all-atom model of VHY required in protein–ligand docking was obtained by adding the hydrogen atoms to all protein atoms. For this purpose, we first determined the protonation states of all ionizable residues according to their hydrogen-bonding patterns revealed in the X-ray structure of VHY. For example, the side chains of Asp and Glu residues were assumed to be neutral if either of their carboxylate oxygens resided in proximity to a hydrogen-bond accepting group within a distance of 3.5 Å, which had been suggested as a distance limit for the hydrogen bonds of moderate strength.11) In the similar way, the side chains of lysine were assumed to be positively charged unless the NZ atom stayed close to a hydrogen-bond donating moiety. This procedure was also applied to assign the protonation states of the side-chain imidazole ring of His residues.
A docking library for VHY was prepared from the latest version of the chemical database (March 2015) distributed by InterBioScreen Ltd. that contained approximately 500000 synthetic and natural molecules. At first, they were filtrated based on Lipinski’s ‘Rule of Five’ to select only the compounds with desirable physicochemical properties as a potential drug candidate12) and with no reactive functional groups. To avoid a structural redundancy among the molecules in the docking library, the similar compounds with Tanimoto coefficient larger than 0.8 were clustered into a single representative one. This two-step filtering resulted in the construction of a docking library comprising approximately 260000 molecules. Three-dimensional atomic coordinates of all these compounds were then generated using the CORINA program with which one can build a stable molecular conformation based on the structural parameters derived from those of known small molecules.13) We finally assigned the atomic charges for all the molecules in the docking library with Gasteiger–Marsilli method14) to compute their electrostatic interaction energies with VHY. The modified version of AutoDock program15) was employed in the molecular docking-based virtual screening of VHY inhibitors. We used the AMBER force field parameters to calculate the conformational energies of putative ligands and the van der Waals interaction energies between VHY and the ligand. Docking simulations were then carried out in the active site VHY to score and rank the molecules in the docking library according to the calculated binding free energies.
In the docking simulations between VHY and a putative inhibitor, we used the empirical scoring function constructed by the substitution of a proper solvation free energy function of ligand in the original AutoDock scoring function, which can be written as follows.
 | (1) |
The DNA fragment coding for human VHY (residues 1–157) was cloned into the pET-28a plasmid (Novagen). The protein was produced in E. coli BL21 (DE3) strain (Novagen). Cells were grown at 18°C overnight after the induction of protein expression with 0.2 mM isopropyl β-D-1-thiogalactopyranoside. Cell pellets were lysed in a buffer containing 50 mM Tris–HCl (pH 7.5), 500 mM NaCl, 1% phenylmethylsulfonyl fluoride (PMSF), 4 mM 2-mercaptoethanol, and 10% glycerol. The protein was purified using Ni-NTA agarose column.
Total 148 compounds selected from the structure-based virtual screening were evaluated by in vitro enzyme assay using the purified VHY protein. These enzyme inhibition assays were performed using 6,8-difluoro-4-methyl-umbelliferyl phosphate (DiFMUP) as a substrate. Purified VHY (30 nM) and DiFMUP (10 µM) were incubated in the reaction mixture containing 100 mM Bis-Tris–HCl (pH 6.0), 200 mM NaCl and 2 mM dithiothreitol for 30 min at 23°C in the presence or absence of an inhibitor candidate. The fluorescence was measured by the PerkinElmer, Inc. 2030 instrument with the excitation and emission wavelengths of 355 and 460 nm, respectively. Biochemical potencies of the identified inhibitors were measured in duplicate at the concentrations of 0, 2, 4, 8, 16, 24, 32, and 40 µM to obtain the dose–response curve fits. IC50 values of the inhibitors were calculated by direct regression analysis using four-parameter sigmoidal curve as implemented in the SigmaPlot program.
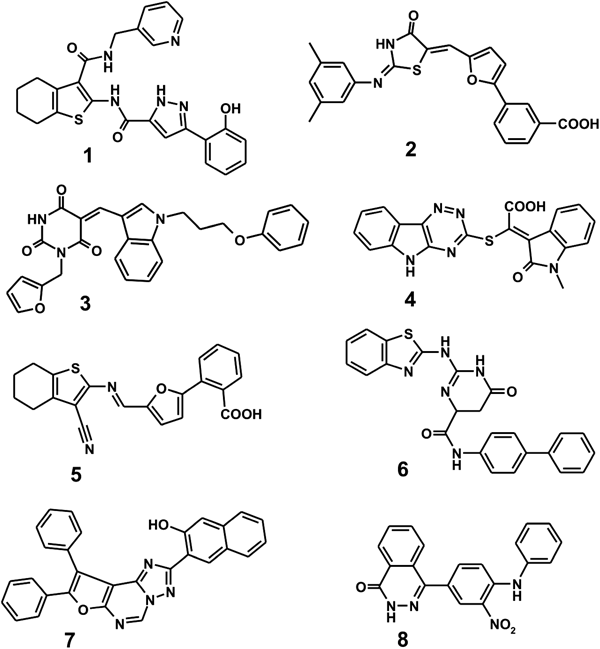
Among more than 260000 compounds screened for the possibility of tight binding in the active site of VHY with the modified AutoDock scoring function, a total of 148 top-scored ones with the calculated ΔGbind values lower than −15 kcal/mol were selected as virtual hits. All these molecules were purchased from the compound supplier and tested for the presence of inhibitory activity against VHY. As the result of extensive enzyme assays, we found eight compounds that inhibited the phosphatase activity of VHY by more than 50% at the concentration of 10 µM, which were selected for further analysis to determine the IC50 values. The chemical structures and IC50 values of the newly discovered VHY inhibitors are shown in Fig. 1 and Table 1, respectively. Actually, none of these eight molecules has been reported as VHY inhibitor so far neither in the literature and nor in patents.
| Compounds | IC50 (µM) | Fluorescence interference (%) |
|---|---|---|
| 1 | 1.8 | 0.3 |
| 2 | 3.3 | 5.1 |
| 3 | 4.1 | 1.7 |
| 4 | 5.2 | 3.4 |
| 5 | 6.2 | 1.5 |
| 6 | 7.1 | 0.9 |
| 7 | 7.5 | 5.8 |
| 8 | 8.7 | 2.6 |
To confirm the biochemical potency as a phosphatase inhibitor, it is required to disprove the possibility that the inhibitor would fluorescently interfere with the assay signal. This validation study started with the incubation of enzymatic reaction mixture for 20 min without the treatment of inhibitor to obtain the maximum of fluorescence signal. At this point, the enzymatic reaction was terminated by the treatment of vanadate, which was followed by the addition of each inhibitor in the reaction mixture to determine its effect of fluorescence interference. The extent of decrease in fluorescence signal was then measured at the inhibitor concentration of 2 µM after the incubation for 5 min. As can be seen in the third column of Table 1, the fluorescence signals appear to be reduced by only 0.3–5.8% due to the interference effects by the inhibitors. These insignificant interference effects indicate that the major contribution to the decrease in fluorescence signal in enzyme assays should come from the inhibition of enzymatic activity rather than from the role of fluorescence quencher played by the inhibitors.
Because compounds 1–8 exhibit good inhibitory activities against VHY and contain several polar moieties as well as the nonpolar aromatic groups, they are likely to be bound tightly in the active site through both the multiple hydrogen bonds and the hydrophobic contacts. It is noteworthy that the eight inhibitors have the low IC50 values ranging from 1 to 9 µM associated with the inhibition of VHY. These micromolar-level inhibitory activities are remarkable because VHY has a flat and shallow active site like other DUSPs in which it is difficult for the inhibitors to be fully accommodated.20) Because 1–8 are structurally diverse and were computationally screened for possessing good physicochemical properties as a drug candidate, each of them deserves consideration for further development by structure–activity relationship (SAR) studies to develop a new medicine for neuroinflammatory diseases.
To gain structural insight into the inhibition of VHY by the identified inhibitors, their binding modes in the active site were examined in a comparative fashion. Figure 2 shows the lowest-energy binding conformations of 1–8 in the active site of VHY obtained with the modified AutoDock scoring function. Docking simulation results for 1–8 are self-consistent because the functional groups with similar chemical character are placed in similar ways to interact with the specific protein groups as revealed by the superimposed structures of 1–8 in Fig. 2. We note in this regard that the polar groups of the inhibitors including 2H-pyrazole-3-carboxylic acid amide (1), carboxylate (2, 4, and 5), pyrimidine-2,4,6-trione (3), 5,6-dihydro-3H-pyrimidin-4-one (6), [1,2,4]triazolo[1,5-c]pyrimidine (7), and nitrobenzene (8) are directed to the active site including the catalytic cysteine residue (Cys88) and hydrogen bond donating groups in the P loop. On the other hand, their nonpolar moieties appear to point toward the residues in P, WPD, and Q loops that reside near the active site. These common features in the calculated binding modes indicate that suitable VHY inhibitors would contain an effective surrogate for the phosphate group in the substrates and simultaneously the nonpolar moieties for binding to the hydrophobic residues in the loop regions. In order to examine the possibility of the allosteric inhibition of VHY by the identified inhibitors, docking simulations were carried out with the grid maps for the receptor model so as to include the entire part of VHY. However, the binding configuration in which an inhibitor resides outside the active site was not observed for any of the new inhibitors. These results support the possibility that the inhibitors would impair the catalytic activity of VHY through the tight binding in the active site.

The positions of P loop, Q loop, WPD loop, and Cys88 are also indicated.
We now turn to the identification of the detailed VHY-inhibitor interactions responsible for the micromolar-level inhibitory activities. The calculated binding mode of 1 in the active site of VHY is shown in Fig. 3. This inhibitor appears to be in close contact with Cys88–Arg94, Asn126–Asn128, and Ala56–Asp57, which belong to PTP, Q and WPD loops, respectively. We note that the sp2 nitrogen on the pyrazole ring and the adjacent aminocarbonyl oxygen of 1 receive a hydrogen bond from the backbone amidic nitrogen of Ser93 and two from the backbone of Ala90 and the side-chain guanidinium ion of Arg94, respectively. These three hydrogen bonds seem to play the role of anchor in positioning the inhibitor in the active site. It is noteworthy that the two hydrogen bond acceptor atoms of 1 reside in the vicinity of the side-chain thiolate ion of Cys88 at a distance within 4.5 Å. Judging from the proximity to Cys88 and the formation of the multiple hydrogen bonds at the bottom of active site, 2H-pyrazole-3-carboxylic acid amide moiety is likely to serve as an effective surrogate for the substrate phosphate group. A stable hydrogen bond is also established between the terminal phenolic group of 1 and the side-chain carboxylate ion of Asp57. The inhibitor 1 can be further stabilized in the active site by the hydrophobic interactions of its nonpolor moieties with the side chains of His38, Ala56, Phe89, Ala90, Ile92, and Pro127. Thus, the overall structural features derived from docking simulations imply that the micromolar inhibitory activity of 1 stems from the multiple hydrogen bonds and hydrophobic interactions established simultaneously in the active site.

Each dotted line indicates a hydrogen bond.
It is also worth noting that three (2, 4, and 5) of the eight micromolar inhibitors found in this study contain the carboxylic acid group. The analysis of the binding mode of 5 is therefore likely to be informative for designing various derivatives with the increased inhibitory activity because of its relatively low molecular weight (376.4 amu). Figure 4 shows the docked pose of 5 that has the lowest free energy of binding in the active site of VHY. The binding mode of 5 differs from that of 1 in that only the backbone amidic groups in the P loop play the roles of hydrogen bond donor with respect to the inhibitor carboxylate ion without the involvement of Arg94. An additional hydrogen bond is also observed between the side-chain amidic nitrogen of Asn126 and the nitrile group of 5, which should also be a significant binding force to stabilize the inhibitor in the active site.

Each dotted line indicates a hydrogen bond.
Hydrophobic interactions in VHY-5 complex appear to be established in the weaker form than in VHY-1 complex: only four aliphatic side chains of Pro59, Ala90, Ile92, and Pro129 of VHY are involved in the van der Waals contacts in the former whereas the aromatic side chains of His38 and Phe89 also contribute to the stabilization of the latter. Because the number of hydrogen bonds is the same in the two VHY-inhibitor complexes, the relatively lower inhibitory activity of 5 than 1 can be attributed to weakening of hydrophobic interactions in the active site. Therefore, the introduction of nonpolar groups at the terminal benzoic acid group of 5 is anticipated to increase the biochemical potency by inducing the new hydrophobic interactions. Because VHY has more hydrophobic residues in the P loop than other DUSPs, such a strengthening of van der Waals interactions in the active site would also be helpful for improving the inhibitor selectivity.
Binding modes of 2–4 and 6–8 are similar to those of 1 and 5 in that an effective surrogate group for the substrate phosphate ion and the nonpolar moieties are stabilized by the multiple hydrogen bonds in the active site around Cys88 and the van der Waals interactions with the hydrophobic residues in P, Q, and WPD loops, respectively (data not shown here). It is thus found to be a common feature in the binding modes of 1–8 that multiple hydrogen bonds and hydrophobic interactions contribute to the stabilization of the inhibitors in the active site in a cooperative fashion.
In summary, we have identified the eight novel inhibitors of VHY by applying a computer-aided drug design protocol involving the molecular docking-based virtual screening under consideration of the effects of ligand solvation on the protein–ligand binding affinity. These inhibitors were also screened in silico for having the desirable physicochemical properties as drug candidate and revealed good biochemical potency with the IC50 values lower than 10 µM. Therefore, each of the newly found inhibitors deserves consideration for further investigation by SAR studies to develop the new medicines for the treatment of neuroinflammatory diseases. The results of binding mode analysis with docking simulations indicated that the VHY inhibitors found in this study would be stabilized in the active site by the simultaneous establishment of multiple hydrogen bonds and van der Waals contacts.
This work was supported by Grants from the National Research Foundation of Korea (2011-0030027) and from the Korea Research Institute of Bioscience and Biotechnology Research Initiative Program.
The authors declare no conflict of interest.