Marine sponge is represented as a considerable source of structurally diverse metabolites with various biological activities, which has attracted a great deal of interest for decades.1) Sponges of genus Diacarnus are recognized as being prolific sources to produce terpene peroxides and related derivatives, which include norterpene ketones, dienes, diols and other types of compounds.2–8) From a biosynthetic perspective, the biosynthetic origin of some or all marine norterpene cyclic peroxides involved a common intermediate C6 hydroperoxide, undergoing a “Michael addition” to an adjacent α,β-unsaturated carboxylic acid or ester moiety.6) As an offshoot of the biosynthetic pathway, the norterpene ketones can be derived from the hydroperoxide carboxylic aid precursor by oxidative degradation.6) Such ketones are increasingly being identified as minor co-metabolites with norterpene cyclic peroxides.3,9–11) Recently, attention has been drawn on these metabolites because of their pharmaceutical properties, including antimalarial,8,12) antimicrobial,12) Trypanosoma brucei inhibitory,13) sea urchin egg cell-division inhibitory,14) cytotoxic,4,7,15) antiulcer and antihypotensive activities.16)
As our previous chemical research on the sponge D. megaspinorhabdosa, five norterpene cyclic peroxides diacarperoxides H–L have been isolated.8) However, to the best of our knowledge, there has been little mention of acyclic terpenes and γ-lactones from the genus of sponge Diacarnus. The interesting metabolites and their bioactive significance of D. megaspinorhabdosa promoted us to study this sponge continuously, which has led to the isolation and identification of two new farnesylacetone derivatives 1–2, a new γ-lactone 3, a known dinorditerpenone 4 and four known norsesterterpene peroxides 5–8 (Fig. 1). Herein, we describe the isolation, structure identification and biological evaluation of these compounds.
Results and DiscussionThe sponge D. megaspinorhabdosa was extracted with ethanol (EtOH) and then partitioned between ethyl acetate (EtOAc) and H2O. The EtOAc phase was divided into two extracts (petroleum ether and CH2Cl2) through solvent-solvent partitioning. The CH2Cl2 phase was fractionated over Sephadex LH-20 to give three subfractions (D1–D3). Subfraction D3 was further subjected to column chromatography (on octadecyl silica (ODS) and silica gel) and semi-preparative HPLC to afford three new compounds 1–3 and five known compounds 4–8. The structures of compounds 1–3 were identified on the basis of spectroscopic techniques, including high resolution-electrospray ionization (HR-ESI)-MS, 1H–1H correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), heteronuclear multiple bond connectivity (HMBC) and nuclear Overhauser effect spectroscopy (NOESY).
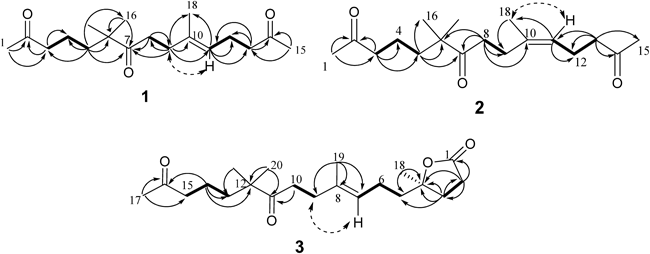
Compound 1 was obtained as a colorless oil. Its molecular formula was established as C18H30O3 by a HR-ESI-MS ion peak at m/z 295.2271 [M+H]+ (Calcd for C18H31O3: 295.2273), which indicating four degrees of unsaturation. The IR spectrum of 1 showed the presence of carbonyl groups (1715 cm−1). The 1H-NMR spectrum displayed five methyls at δH 1.60 (3H, s), 1.09 (6H, s), 2.10 (3H, s), 2.11 (3H, s) and a olefinic proton at δH 5.07 (1H, t, J=6.1 Hz). The 13C-NMR and distortionless enhancement by polarization transfer (DEPT) spectra of 1 revealed the presence of 18 carbon signals consisting of five methyls, seven methylenes, one methine and five quaternary carbons. Three ketones functional groups (δC 208.4, 208.6, and 214.8), and a double bond (δC 122.9, 135.3) were attributed to four degrees of unsaturation, which indicated compound 1 was acyclic. Further examination of the 1D NMR spectra (Table 1) data revealed a single trisubstituted double bond (δH 5.07, δC 122.9), (δC 135.3), two acetyls (δH 2.10, δC 29.8, δC 208.4), (δH 2.11, δC 29.8, δC 208.6), one olefinic methyl (δH 1.60, δC 16.0), two methyl singlets (both δH 1.09, δC 24.2). Analysis of the COSY data allowed the assembly of the H2-3 (δH 2.39)/H2-4 (δH 1.40)/H2-5 (δH 1.44), H2-8 (δH 2.51)/H2-9 (δH 2.18), and H-11 (δH 5.07)/H2-12 (δH 2.22)/H2-13 (δH 2.43) units (Fig. 2). Assignment and connection between these units were supported by the HMBC experiment. The HMBC correlations from H2-5 to C-3 (δC 43.8), C-4 (δC 18.9), C-6 (δC 47.4), C-7 (δC 214.8), C-16 (δC 24.2), and C-17 (δC 24.2) together with correlations from H2-9 to C-7 (δC 214.8), C-8 (δC 35.4), C-10 (δC 135.3), C-11 (δC 122.9), and C-18 (δC 16.0) established the connectivity of the partial structure C-3 to C-13. The initial methyl CH3-1 (δH 2.10) showed the HMBC correlations to C-2 (δC 208.4) and C-3 (δC 43.8), and terminal methyl CH3-15 (δH 2.11) displayed correlations to C-14 (δC 208.6), and C-13 (δC 43.4), which supported the assignment of the methyls attached to ketones as shown in Fig. 2.
Table 1.
1H- (600 MHz) and
13C-NMR (150 MHz) Data for
1–
3 in CDCl
3| No. | 1 | 2 | 3 |
|---|
| δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) |
|---|
| 1 | 29.8 (q) | 2.10 (s) | 29.9(q) | 2.14 (s) | 176.7 (s) | |
| 2 | 208.4 (s) | | 208.5 (s) | | 29.1 (t) | 2.62 (m) |
| 3 | 43.8 (t) | 2.39 (t, 7.1) | 43.9 (t) | 2.43 (t, 6.6) | 33.0 (t) | 1.99 (m); 2.01 (m) |
| 4 | 18.9 (t) | 1.40 (m) | 19.0 (t) | 1.49 (m) | 86.6 (s) | |
| 5 | 39.0 (t) | 1.44 (m) | 39.2 (t) | 1.45 (m) | 40.8 (t) | 1.68 (m); 1.72 (m) |
| 6 | 47.4 (s) | | 47.4 (s) | | 22.5 (t) | 2.08 (m) |
| 7 | 214.8 (s) | | 214.8 (s) | | 123.6 (d) | 5.12 (t, 7.2) |
| 8 | 35.4 (t) | 2.51 (t, 7.4) | 35.2 (t) | 2.54 (t, 7.6) | 135.1 (s) | |
| 9 | 33.3 (t) | 2.18 (t, 7.4) | 26.0 (t) | 2.26 (d, 7.6) | 33.4 (t) | 2.20 (t, 7.8) |
| 10 | 135.3 (s) | | 135.3 (s) | | 35.5 (t) | 2.55 (t, 7.8) |
| 11 | 122.9 (d) | 5.07 (t, 6.1) | 124.2 (d) | 5.10 (t, 7.5) | 214.9 (s) | |
| 12 | 22.3 (t) | 2.22 (quin, 7.5) | 22.2 (t) | 2.28 (t, 7.3) | 47.5 (s) | |
| 13 | 43.4 (t) | 2.43 (t, 7.5) | 43.8 (t) | 2.47 (t, 7.3) | 39.2 (t) | 1.48 (m) |
| 14 | 208.6 (s) | | 208.6 (s) | | 19.0 (t) | 1.44 (m) |
| 15 | 29.8 (q) | 2.11 (s) | 29.9 (q) | 2.15 (s) | 43.9 (t) | 2.42 (t, 6.6) |
| 16 | 24.2 (q) | 1.09 (s) | 24.3 (q) | 1.14 (s) | 208.5 (s) | |
| 17 | 24.2 (q) | 1.09 (s) | 24.3 (q) | 1.14 (s) | 29.9 (q) | 2.13, (s) |
| 18 | 16.0 (q) | 1.60 (s) | 23.2 (q) | 1.68 (brs) | 25.6 (q) | 1.41, (s) |
| 19 | | | | | 16.1 (q) | 1.62, (s) |
| 20 | | | | | 24.3 (q) | 1.13, (s) |
| 21 | | | | | 24.3 (q) | 1.13, (s) |
The Δ10,11 double bond in 1 was established on the basis of NOESY experiment. The E-geometry of this double bond was deduced from the carbon chemical shift of the C10-methyl (δC 16.0, C-18)7,17) and the NOESY correlation between H-9 (δH 2.18) and H-11 (δH 5.07) (Fig. 2). Therefore, the new compound 1 was determined as (E)-6,6,10-trimethylpentadec-10-ene-2,7,14-trione.
Compound 2 was also isolated as a colorless oil, with a molecular formula as C18H30O3 determined by its HR-ESI-MS (m/z 295.2274 [M+H]+, Calcd for C18H31O3: 295.2273) and 13C-NMR data. Four degrees of unsaturation implied by the molecular formula were assigned to three ketones functional groups (δC 208.5, 208.6, and 214.8), and a double bond (δC 124.2, 135.3). The IR spectrum displayed band at 1712 cm−1 due to carbonyl groups. The 1H-NMR spectrum exhibited five methyls at δH 1.68 (3H, br s), δH 1.14 (6H, s), δH 2.14 (3H, s), δH 2.15 (3H, s) and a olefinic proton at δH 5.10 (1H, t, J=7.5 Hz). The 13C-NMR and DEPT spectra of 2 revealed the presence of 18 carbon signals consisting of five methyls, seven methylenes, one methine and five quaternary carbons. Comparison of the 1D NMR spectra of compounds 1 with those of 2 (Table 1) unequivocally revealed that these two compounds had similar planar structures, which was confirmed by the extensive 2D-NMR spectroscopic analysis. The COSY correlations between H-11 (δH 5.10)/H-12 (δH 2.28)/H-13 (δH 2.47), H-8 (δH 2.54)/H-9 (δH 2.26), together with the HMBC correlations from H-13 to C-11 (δC 124.2), C-12 (δC 22.2), C-14 (δC 208.6), and C-15 (δC 29.9), and from H-8 to C-6 (δC 47.4), C-7 (δC 214.8), C-9 (δC 26.0), and C-10 (δC 135.3) completed the assignment of compound 2 (Fig. 2, Table 1). The geometry of double (Δ10,11) was determined as Z on the basis of the NOESY correlation between H-11 (δH 5.10) and H-18 (δH 1.68), and was also confirmed by the comparison of the chemical shift of C-18 (δC 23.2) with the literature (δC 20–25 for Z, δC 12–17 for E).17) Consequently, the new compound 2 was established as (Z)-6,6,10-trimethylpentadec-10-ene-2,7,14-trione.
Compound 3 was obtained as a colorless oil. The molecular formula was determined to be C21H34O4 by HR-ESI-MS (m/z 373.2352 [M+Na]+, Calcd for C21H34O4Na: 373.2355). The IR spectrum showed bands at 1770 cm−1 and 1708 cm−1 due to carbonyl groups. Five degrees of unsaturation implied by the molecular formula were assigned to a ring, three ketone functional groups (δC 176.7, 208.5, and 214.9), and a double bond (δC 123.6 and 135.1). The 1H-NMR spectrum displayed five methyls at δH 1.13 (6H, s), 1.41 (3H, s), 1.62 (3H, s), and 2.13 (3H, s) and a olefinic proton at δH 5.12 (1H, t, J=7.2 Hz). The 13C-NMR and DEPT spectra of 3 revealed the presence of 21 carbon signals consisting of five methyls, nine methylenes, one methine and six quaternary carbons. Comparison of the 1D NMR spectroscopic data of 3 with those of 1 disclosed that these two compounds shared the same (E)-6,6,10-trimethyldodec-10-ene-2,7-dione moiety. Additionally, the HMBC correlations from H2-2 (δH 2.62) to C-1 (δC 176.7), C-3 (δC 33.0), and C-4 (δC 86.6), from H2-3 (δH 1.99, 2.01) to C-1 (δC 176.7), C-2 (δC 29.1), C-4 (δC 86.6), C-5 (δC 40.8), and C-18 (δC 25.6) suggested the presence of a γ-lactone group, which was also confirmed by the carbon resonances at C-4 (δC 86.6) and C-1 (δC 176.7).10) Consequently, 3 was suggested to be composed of a (E)-6,6,10-trimethyldodec-10-ene-2,7-dione moiety linked through a C2H4 chain to the γ-lactone group. This assignment was confirmed by the COSY correlations between H2-5 (δH 1.68, 1.72) and H2-6 (δH 2.08), H2-6 and H-7 (δH 5.12). The geometry of the double bond was assigned as E through the NOESY correlation between H-7 (δH 5.12) and H-9 (δH 2.20), which was also confirmed by comparison of the chemical shift of C-19 (δC 16.1) with literature.17) Its circular dichroism (CD) spectrum exhibited a positive cotton effect (Δε+0.15) at 217 nm attributed to the n→π* electronic transition of the γ-lactone.18,19) Therefore, the absolute configuration at C-4 was assigned as R.
Besides the three new compounds 1–3, the known compounds 4–8 were identified as muquketone (4),3) aikupikoxide A (5),4) (−)-ent-muqubilone methyl ester (6),13) muqubilin (7),14) (+)-epimuqubilin A methyl ester (8)3) by the comparison of their spectroscopic data with those in the literatures.
The cytotoxicity of compounds 1–8 against five human cancer cell lines including NCI-H446 (small-cell lung cancer), NCI-H440 (large-cell lung cancer), SGC-7901 (gastric carcinoma), HeLa (cervical cancer), and MCF-7 (breast carcinoma) were tested by CCK-8 assay. Inhibition of cell proliferation by these compounds at various concentrations were measured, and their IC50 values were calculated. The results showed that compound 3 exhibited moderate cytotoxicity against the above five cancer cell lines with IC50 values of 18.5, 20.1, 22.3, 26.7, and 47.1 µM, respectively. The other tested compounds were not cytotoxic (IC50>50 µM) toward the five cancer cell lines.
ExperimentalGeneral Experimental ProceduresAll NMR spectra were recorded on Bruker AVANCE-600 spectrometers (600 and 150 MHz for 1H- and 13C-NMR, respectively) in CDCl3. Chemical shifts were reported in ppm referenced to CHCl3 as internal standard (δ 7.26 for proton and δ 77.0 for carbon). Optical rotation was determined with a Perkin-Elmer model 341 polarimeter with 1 mm cell. The CD spectra was recorded on a JASCO J-715 spectropolarimeter. HR-ESI-MS and ESI-MS spectra were obtained on a Waters Q-Tof micro YA019 mass spectrometer. IR spectra were recorded on a Bruker vector 22 spectrometer with KBr pellets. Reversed-phase HPLC were performed on YMC-Pack Pro C18 RS columns (250×10 mm, 5 µm) using a Waters 1525/2998 high performance liquid chromatography. Column chromatography (CC) was performed on Sephadex LH-20 (Amersham Biosciences). Vacuum liquid chromatography (VLC) was carried out using silica gel 60 (200×300 mesh, Qingdao Ocean Chemical Company, China); the fractions were monitored by TLC (HSGF 254, Yantai, China), and spots were visualized by heating silica gel plates sprayed with 10% anisaldehyde–H2SO4 reagent.
Animal MaterialThe sponge was collected around Yongxing Island and Seven Connected Islets in the South China Sea in April 2010. The sample was directly frozen after collection and stored at −10°C. It was kindly identified by Prof. Jin-He Li (Institute of Oceanology, Chinese Academy of Sciences, China). A voucher specimen (No. XD-2010008) was deposited in the Laboratory of Marine Drugs, Department of Pharmacy, Changzheng Hospital, Second Military Medical University, China.
Extraction and IsolationThe freeze-dried sponge (1.8 kg, dry weight) was extracted three times (3×50 L) with 95% aqueous EtOH and the combined extracts were concentrated under reduced pressure at 45°C to obtain the crude extract, which was redissolved in H2O (500 mL). The aqueous solution was extracted with EtOAc (3×500 mL) and n-BuOH to afford the EtOAc-soluble (44.2 g) and n-BuOH-soluble extracts. The EtOAc-soluble extract (44.2 g) was partitioned between 90% aqueous methanol (MeOH) (300 mL) and petroleum ether (3×300 mL) to yield the petroleum ether-soluble fraction (29.1 g). The 90% MeOH/H2O phase was diluted to 60% MeOH/H2O and extracted with CH2Cl2 to afford the CH2Cl2-soluble fraction (13.7 g), which was fractionated over Sephadex LH-20 eluted with n-hexane/dichloromethane/methanol (4 : 5 : 1) to give three subfractions (D1–D3). Subfraction D3 (10.4 g) was subjected to vacuum liquid chromatography (VLC) on silica gel by gradient elution using petroleum ether/ethyl acetate (stepwise, 100 : 1, 50 : 1, 20 : 1, 15 : 1, 10 : 1, 5 : 1, 1 : 1, 0 : 1), to give eight subfractions (D3A–D3H). Fraction D3D (1.48 g) was subjected to CC on ODS and further purified by reversed-phase preparative HPLC (YMC-Pack Pro C18 RS, 5 μm, 10×250 mm, 2.0 mL/min), to obtain compound 1 (CH3CN/H2O 55 : 45, 2.0 mL/min, UV detection at 200 nm, tR=22.2 min, 20.0 mg), compound 2 (CH3CN/H2O 45 : 55, 2.0 mL/min, UV detection at 195 nm, tR=47.8 min, 2.1 mg), compound 3 (CH3CN/H2O 45 : 55, 2.0 mL/min, UV detection at 195 nm, tR=62.6 min, 1.8 mg), and compound 8 (CH3CN/H2O 85 : 15, 2.0 mL/min, UV detection at 200 nm, tR=122.5 min, 3.9 mg). Fraction D3C (500 mg) was separated by silica gel column chromatography eluted with CH2Cl2/MeOH (20 : 1–1 : 1, v/v) to afford compound 5 (10.4 mg) and compound 7 (30.0 mg). Fraction D3B (947 mg) was purified using a Sephadex LH-20 column eluted with CH2Cl2/MeOH (1 : 1, v/v) and followed by HPLC (YMC-Pack Pro C18 RS, 5 μm, 10×250 mm, 2.0 mL/min) to yield 4 (CH3CN/H2O 90 : 10, 2.0 mL/min, UV detection at 200 nm, tR=30.8 min, 3.0 mg) and 6 (CH3CN/H2O 85 : 15, 2.0 mL/min, UV detection at 200 nm, tR=51.2 min, 20.0 mg).
(E)-6,6,10-Trimethylpentadec-10-ene-2,7,14-trione (1)Colorless oil; UV (CH3OH), λmax (log ε): 207 (3.26) nm; IR (KBr) cm−1: 3504, 3410, 2965, 2931, 1715, 1471, 1435, 1410, 1362, 1264, 1231, 1176, 1161, 1103, 1080, 986, 864, 726, 600, 519. 1H-NMR (CDCl3, 600 MHz) and 13C-NMR (CDCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 295.2271 [M+H]+ (Calcd for C18H31O3, 295.2273).
(Z)-6,6,10-Trimethylpentadec-10-ene-2,7,14-trione (2)Colorless oil; UV (CH3OH), λmax (log ε): 209 (3.38) nm; IR (KBr) cm−1: 3446, 2964, 2927, 2858, 1712, 1453, 1412, 1362, 1260, 1163, 1080, 728. 1H-NMR (CDCl3, 600 MHz) and 13C-NMR (CDCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 295.2274 [M+H]+ (Calcd for C18H31O3, 295.2273).
Diacarlactone A (3)Colorless oil; [α]D27+2.07 (c 0.12, CH3OH); CD (3.4×10−3 M, CH3OH) λmax (Δε) 217 (+0.15). UV (CH3OH), λmax (log ε): 210 (3.80) nm; IR (KBr) cm−1: 3471, 2961, 2928, 2858, 1770, 1708, 1459, 1416, 1380, 1363, 1281, 1259, 1201, 1170, 1085, 1025, 937, 802, 727, 663, 536. 1H-NMR (CDCl3, 600 MHz) and 13C-NMR (CDCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 373.2352 [M+Na]+ (Calcd for C21H34O4Na: 373.2355).
CCK-8 Cytotoxicity AssayThe CCK-8 method20) was used for in vitro evaluation of the cytotoxic potential of compound 1–8 against five human cancer cell lines including HeLa, NCI-H446, NCI-H460, SGC-7901, and MCF-7. All the cells were cultured in RPMI-1640 or Dulbecco’s modified Eagle’s medium (DMEM) medium (Hyclone), supplemented with 10% fetal bovine serum (Hyclone) in 5% CO2 at 37°C. The cytotoxicity assay was performed in 96-well microplates. Briefly, Cells in the exponential phase were seeded in 96-well culture plates at the confluence of 5×103 cells/well, kept in 37°C, 5% CO2 incubator for 24 h. Replaced the medium containing different concentrations of compound in fresh medium at the same conditions incubator for 48 h with cisplatin as positive control, and then added 90 µL of fresh medium and 10 µL CCK-8, kept at 37°C and 5% CO2 for 1h. Absorption was then measured by using a SpectraMAX 340 reader (Molecular Devices, Sunnyvale, CA, U.S.A.) at 450 nm and IC50 values were calculated on the basis of percentage of inhibition using the linear regression method. The experiments were carried out in triplicate (n=3), and DDP (cis-dichlorodiamineplatinum (II), Sigma) was chosen as positive control.