Results and Discussion
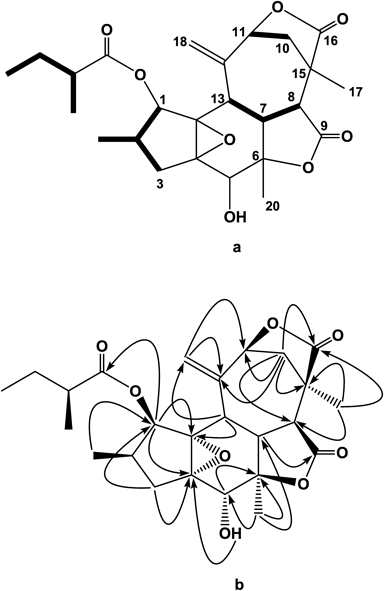
A CH2Cl2-soluble fraction of the MeOH extract of the stems of C. cascarilloides was separated by various kinds of chromatography to give six new crotofolane diterpenoids, named crotocascarins L–Q (1–6), and a diterpenoid with a new scaffold, named neocrotocascarin (7).
Crotocascarin L (1), [α]D24 +6.8, was isolated as an amorphous powder and its elemental composition was determined to be C22H26O7 by observation of a quasi-molecular ion peak ([M+Na]+) at m/z: 425.1562 on high-resolution (HR)-electrospray ionization (ESI)-MS. The IR spectrum showed a strong absorption band at 1747 cm−1 assignable to that of an α,β-unsaturated γ-lactone. The 13C-NMR spectrum exhibited 22 resonances including that of one acetyl group (δC 20.6, 169.5) (Table 1), the remaining 20 signals being essentially the same as those of crotocascarin A.2) An obvious downfield shift was observed for H-1 (δH 5.47), to which an acetoxy group was expected be attached (Table 2). This was confirmed by the heteronuclear multiple bond correlation (HMBC) from H-1 to the carbonyl carbon in the acetoxy moiety. Therefore, the structure of 1 was elucidated to be a 1-O-acetyl derivative of crotocascarin A, as shown in Fig. 1.
Table 1.
13C-NMR Spectroscopic Data for Crotocascarins L–Q (
1–
6) and Neocrotocascarin (
7) (CDCl
3, 100 MHz)
| C | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|
| 1 | 74.9 | 73.7 | 209.7 | 75.7 | 75.1 | 75.0 | 74.9 |
| 2 | 33.4 | 33.6 | 36.8 | 32.8 | 32.4 | 32.4 | 32.9 |
| 3 | 36.9 | 36.3 | 34.1 | 37.0 | 37.3 | 36.0 | 35.3 |
| 4 | 60.5 | 60.3 | 60.8 | 66.5 | 66.4 | 67.6 | 65.9 |
| 5 | 57.8 | 58.2 | 58.0 | 72.7 | 74.2 | 70.9 | 73.9 |
| 6 | 56.4 | 56.6 | 57.2 | 75.5 | 75.4 | 145.5 | 85.3 |
| 7 | 44.1 | 44.1 | 44.8 | 41.9 | 41.7 | 39.2 | 50.3 |
| 8 | 158.9 | 159.7 | 158.0 | 158.8 | 162.9 | 162.7 | 51.1 |
| 9 | 107.8 | 109.7 | 107.3 | 106.6 | 79.9 | 78.5 | 170.7 |
| 10 | 42.0 | 35.8 | 41.9 | 47.7 | 44.4 | 42.9 | 45.8 |
| 11 | 35.0 | 34.9 | 34.7 | 73.6 | 72.5 | 73.2 | 78.4 |
| 12 | 146.6 | 147.7 | 146.6 | 147.5 | 148.6 | 148.9 | 147.7 |
| 13 | 39.6 | 39.5 | 38.4 | 29.3 | 29.5 | 35.9 | 30.9 |
| 14 | 68.8 | 70.7 | 66.2 | 71.2 | 71.7 | 71.4 | 68.4 |
| 15 | 130.7 | 129.8 | 130.8 | 130.2 | 127.0 | 127.0 | 40.7 |
| 16 | 170.4 | 170.9 | 170.3 | 170.1 | 174.0 | 173.7 | 178.1 |
| 17 | 9.7 | 9.6 | 9.7 | 11.2 | 11.2 | 9.6 | 24.6 |
| 18 | 115.1 | 114.6 | 113.4 | 116.5 | 116.0 | 115.5 | 114.1 |
| 19 | 12.5 | 12.2 | 12.9 | 12.3 | 12.3 | 12.3 | 12.7 |
| 20 | 20.2 | 20.3 | 20.0 | 24.4 | 24.2 | 114.0 | 22.8 |
| 1′ | 169.5 | | | 178.9 | 178.0 | 178.2 | 175.8 |
| 2′ | 20.6 | | | 41.5 | 41.4 | 41.3 | 41.0 |
| 3′ | | | | 26.2 | 26.2 | 26.4 | 26.7 |
| 4′ | | | | 11.6 | 11.6 | 11.5 | 11.6 |
| 5′ | | | | 16.7 | 16.5 | 16.5 | 16.6 |
| –OCH3 | | 51.6 | | | | | |
Table 2.
1H-NMR Spectroscopic Data for Crotocascarins L–Q (
1–
6) and Neocrotocascarin (
7) (CDCl
3, 400 MHz)
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|
| 1 | 5.47 d 6 | 4.19 dd 5, 5 | | 5.38 d 5 | 5.40 d 5 | 5.37 d 5 | 5.59 d 4 |
| 2 | 2.20 m | 2.05 m | 2.52 m | 2.13 m | 2.13 m | 2.18 m | 2.15 m |
| 3 | 2.47 dd 14, 7 | 2.41 dd 14, 7 | 2.82 dd 14, 8 | 2.45 dd 14, 7 | 2.45 dd 13, 7 | 2.49 dd 14, 7 | 2.43 dd 14, 7 |
| 1.65 dd 14, 10 | 1.65 dd 14, 10 | 1.62 dd 14, 8 | 1.66 dd 14, 10 | 1.61 dd 13, 10 | 1.60 dd 12, 4 | 1.55 dd 14, 10 |
| 5 | 3.16 s | 3.10 s | 3.09 s | 3.71 br s | 3.61 d 8 | 4.48 s | 4.38 d 7 |
| 7 | 2.99 dq 13, 1 | 2.97 dq 13, 1 | 2.88 d 13 | 2.97 d 11 | 2.81 d 11 | 3.52 d 11 | 2.44 dd 11, 9 |
| 8 | | | | | | | 3.15 d 9 |
| 9 | | | | | 5.21 o | 5.28 br d 12 | |
| 10 | 2.46 o | 2.81 ddd 14, 7, 7 | 2.45 o | 2.74 dd 15, 4 | 2.63 ddd 14, 5, 5 | 2.74 ddd 14, 4, 4 | 2.46 dd 13, 8 |
| 1.59 ddd 13, 13, 5 | 1.37 ddd 14, 14, 4 | 1.53 m | 1.72 dd 15, 4 | 1.36 ddd 14, 9, 4 | 1.34 ddd 14, 12, 4 | 2.08 br d 13 |
| 11 | 2.46 o | 2.45 ddd 14, 4, 4 | 2.43 o | 4.47 dd 4, 4 | 4.33 br s | 4.52 dd 4, 4 | 5.03 br d 8 |
| 2.11 ddd 13, 13, 5 | 2.15 ddd 14, 14, 4 | 2.43 o | | | | |
| 13 | 3.05 d 13 | 3.26 d 13 | 3.09 d 13 | 3.97 d 11 | 3.72 d 11 | 3.01 d 11 | 3.03 d 11 |
| 17 | 1.91 3H s | 1.89 3H d 1 | 1.85 3H s | 1.79 3H, br s | 1.77 3H d 1 | 1.67 3H br s | 1.74 3H s |
| 18 | 5.08 s | 5.07 s | 4.94 s | 5.35 s | 5.33 s | 5.41 s | 5.29 s |
| 5.06 br s | 5.05 br s | 4.89 s | 5.26 br s | 5.23 br s | 5.24 s | 5.26 s |
| 19 | 0.92 3H d 7 | 1.03 3H d 7 | 1.07 d 7 | 0.94 3H d 7 | 0.93 3H d 7 | 0.94 3H d 7 | 0.88 3H d 7 |
| 20 | 1.18 3H s | 1.12 3H s | 1.10 3H s | 1.15 3H s | 1.19 3H s | 5.13 s | 1.45 3H s |
| | | | | | 4.84 d 1 | |
| 2′ | | | | 2.45 qt 7, 7 | 2.44 qt 7, 7 | 2.41 qt 7, 7 | 2.43 o |
| 3′ | | | | 1.73 dqd 14, 7, 7 | 1.76 dqd 14, 7, 7 | 1.70 dqd 14, 7, 7 | 1.69 dqd 14, 7, 7 |
| | | | 1.45 dqd 14, 7, 7 | 1.46 dqd 14, 7, 7 | 1.45 dqd 14, 7, 7 | 1.44 dqd 14, 7, 7 |
| 4′ | | | | 0.91 t 7 | 0.93 3H t 7 | 0.90 3H t 7 | 0.90 3H t 7 |
| 5′ | | | | 1.18 3H d 7 | 1.20 3H d 7 | 1.14 3H d 7 | 1.17 3H d 7 |
| 1-OH | | 1.47 d 5 | | | | | |
| 5-OH | | | | | 2.38 d 8 | | 2.23 d 7 |
| 9-OH | 3.43 br s | | | | | | |
| 11-OH | | | | | 2.31 br s | | |
| CH3CO– | 2.11 3H s | | | | | | |
| CH3O– | | 3.52 3H s | | | | | |
Crotocascarin M (2), [α]D24 +44.2, was isolated as colorless needles and its elemental composition was determined to be C21H26O6 by HR-ESI-MS. The spectroscopic data showed close resemblance with those of crotocascarin J, whose hemiketal carbon (C-9) appeared at δC 108.0 in the 13C-NMR spectrum.3) Similarly, the ketal carbon of 2 appeared at δC 109.7 along with methoxy signals at δH 3.52 and δC 51.6 in the NMR spectra, and the methoxy protons crossed the ketal carbon (δC 109.7) in the HMBC spectrum. Therefore, the structure of 2 was elucidated to be a methoxy derivative of crotocascarin J, as shown in Fig. 1.
Crotocascarin N (3), [α]D26 +2.8, was isolated as an amorphous powder and its elemental composition was determined to be C20H22O6 by HR-ESI-MS. In the IR spectrum, a strong absorption band (1739 cm−1) for carbonyl groups was observed. NMR revealed that 3 was a similar compound to 2, except for a hydroxy functional group at the 9-position, and the oxygenated methine signal (δC 73.7) (C-1) in 2 appeared at δC 209.7 as a carbonyl group. The position of this carbonyl group was confirmed by the HMBC correlation cross peaks from H3-19 (δH 1.07) and H-13 (δH 3.09) (JC-C-C-H=4 Hz) to C-1. Therefore, the structure of 3 was elucidated, as shown in Fig. 1.
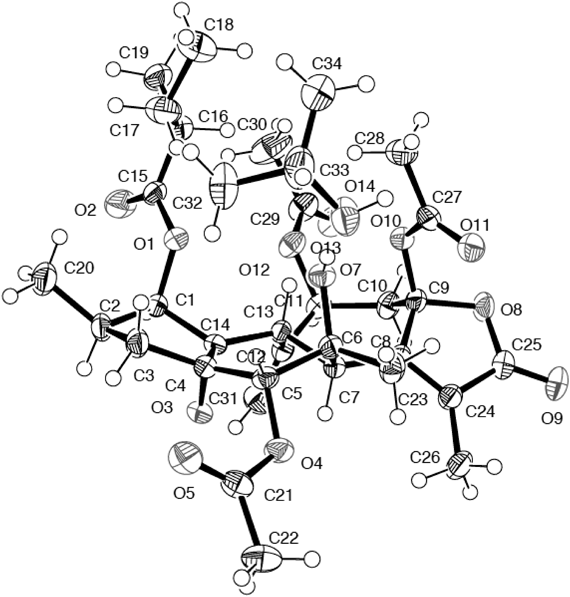
Crotocascarin O (4), [α]D25 +103.1, was isolated as an amorphous powder and its elemental composition was determined to be C25H34O9 by HR-ESI-MS. In the IR spectrum, an absorption band assignable to an α,β-unsaturated γ-lactone was observed at 1743 cm−1 and the 13C-NMR spectrum showed 25 resonances. Of the 25 13C-NMR signals, five were assigned to those of 2-methylbutyric acid and the remaining 20 signals were similar to those of 1, comprising those of three methyls, two methylenes, di- and tetrasubstituted double bonds, three methines, three oxygenated methines, three oxygenated tertiary carbons, one ketal and carbonyl carbon signals. The fairly shielded oxymethine (C-5, δC 57.8) and oxygenated tertiary carbon (C-6, δC 56.4) signals seen for 1 were shifted downfield to δC 72.7 and 75.5, respectively (Table 1), with simultaneous loss of one degree of unsaturation, compared with those for 1. These findings indicated that the opening of an epoxy ring between C-5 and C-6 had taken place, and the C-5 and C-6 vicinal diol system must be elaborated. To confirm the geometry of these hydroxy groups, X-ray crystallographic analysis of crotocascarin N 5,9,11-tri-O-acetate (4a) was performed and an ORTEP drawing is presented in Fig. 2. The absolute configuration was expected to be the same as those of other crotofolanes isolated from C. cascarilloides from the positive Cotton effect at 254 nm in the CD spectrum,2,8,9) and chirality analysis of the 2-methylbutanoic acid moiety by HPLC with an optical rotation detector unambiguously established the absolute configuration of 4 to be shown as in Figs. 1 and 2.
Crotocascarin P (5), [α]D25 +96.9, was isolated as an amorphous powder and its elemental composition was determined to be C25H34O8 by HR-ESI-MS. Crotocascarin P (5) was an analogous compound to 4 in which the hemiketal carbon was replaced by an oxymethine one. The positive Cotton effect at 260 nm in the CD spectrum also supported the absolute structure was the same as that of 4. Therefore, the structure of 5 is as shown in Fig. 1.
Crotocascarin Q (6), [α]D25 +146.2, was isolated as an amorphous powder and its elemental composition was determined to be C25H32O7. In the 13C-NMR spectrum, five typical signals assignable to 2-methyl butyric acid and two sets [δC 114.0 (t), 145.5 (s), 115.5 (t), 148.9 (s)] of exo-cyclic methylene signals were observed. Two sets of exo-cyclic methylene signals are a characteristic of crotocascarin K (8),3) also with a β-hydroxy group at the 5-position and an epoxy group between C-4 and C-14, and an extra hydroxy group at the 11-position. When an α,β-unsaturated γ-lactone with a parallel double bond exists at the close region, the empirical γ-lactone rule8,9) cannot be adopted for determination of the absolute structure. Since the absolute structure of 8 was independently determined by a combination of X-ray crystallographic analysis and the modified Mosher’s method,3) a similar CD Cotton effect at 224 nm (∆ε+17.2) to that of crotocascarin K at 226 nm (∆ε+17.2) established that the absolute stereochemistry at the 9-position of 6 was the same as that of 8. The hydroxy group at the 11-position was expected to be on the β-side from the similar coupling pattern of H-11 to that in 4. Therefore, the structure of 6 was elucidated to be as shown in Fig. 1.
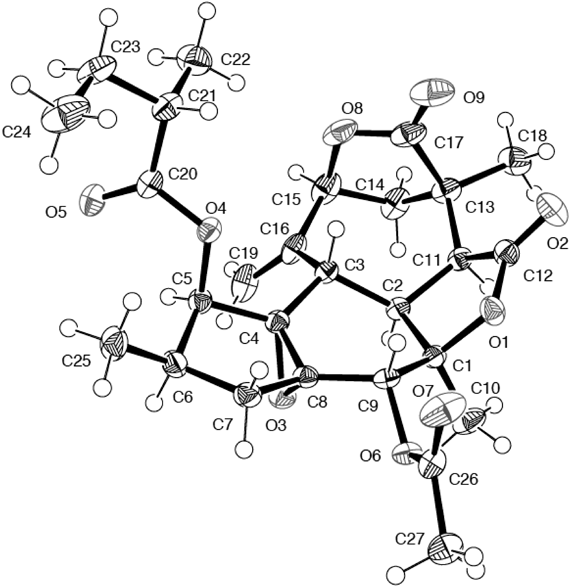
Neocrotocascarin (7), [α]D24 +5.0, was isolated as an amorphous powder and its elemental composition was determined to be C25H32O8 by HR-ESI-MS. In the IR spectrum, absorptions assignable to γ-lactone (1776 cm−1) and an ester carbonyl group (1744 cm−1) were observed. NMR spectra indicated the presence of a 2-methylbutyric acid moiety, and one doublet and two singlet methyl signals were observed in the 1H-NMR spectrum. Two sp2 carbon signals [δC 147.7 (s) and 114.1 (t) with δH 5.26 and 5.29] observed in the NMR spectrum implied an exo-cyclic methylene which existed in co-occurring crotofolanes (1–6), must undergo some modification. This implication was also suggested by the proton sequence from H-8 to H-7 and then to H-13 on the 1H–1H correlation spectroscopy (COSY) (Fig. 3a), and a HMBC correlation from H-10 (δH 2.08) to C-16 (δC 178.1) (Fig. 3b). Acetylation of 7 gave its monoacetate (7a) and this monoacetate was crystallized from n-hexane–CHCl3 to give a suitable crystal for X-ray analysis. An ORTEP drawing of 7a is shown in Fig. 4 and the structure was proved to have a new skeleton. The acyl moiety was found on HPLC using a chiral detector to have a positive rotation sign, namely the S-form. Therefore, the structure of neocrotocascarin (7) including its absolute configuration was elucidated to be as shown in Fig. 1.
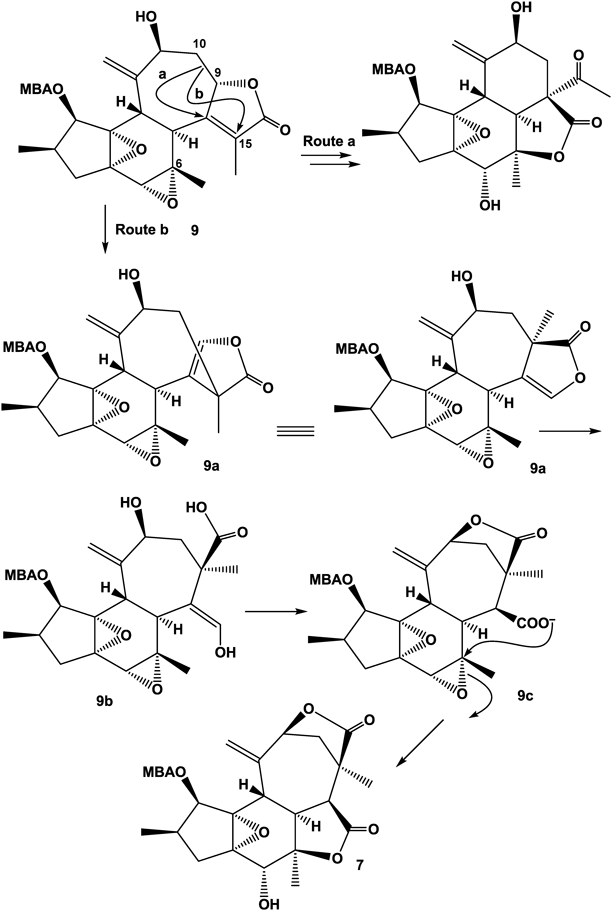
A plausible biosynthetic mechanism for the new skeleton is summarized in Fig. 5. In a typical crotofolane (9), the C-9–C-10 bond migrates to C-8 (route a) and then proceeds to rearranged nor-crotofolane, crotocascarins α–γ series.1,2) While the C-9–C-10 bond migration through route b gives compound 9a. After the lactone ring opening (9b), re-formation of a lactone ring with the hydroxy group at C-11 leads to 9c. The aldehyde equivalent vinyl alcohol of 9c is then oxidized and the formed carboxylic anion attacks C-6 to open an epoxide ring, simultaneously forming another γ-lactone to give a new skeleton, neocrotocascarin.
Experimental
General Experimental ProcedureMelting points were measured on a Yanagimoto micro melting point apparatus and are uncorrected. Optical rotations were measured on a JASCO P-1030 digital polarimeter. IR and UV spectra were measured on Horiba FT-710 and JASCO V-520 UV/Vis spectrophotometers, respectively. 1H- and 13C-NMR spectra were taken on a JEOL JNM α-400 at 400 and 100 MHz, respectively, with tetramethylsilane as an internal standard. CD spectra were obtained with a JASCO J-720 spectropolarimeter. Positive-ion HR-ESI-MS was performed with an Applied Biosystems QSTAR XL NanoSprayTM System. Silica gel column chromatography (CC) was performed on Kiesel Gel (silica gel 60) (70–230 mesh) (E. Merck, Darmstadt, Germany), and reversed-phase octadecylsilanized (ODS) open CC on Cosmosil 75C18-OPN (Nacalai Tesque, Kyoto, Japan) (Φ=50 mm, L=25 cm). HPLC was performed on an ODS column (Inertsil ODS-3 and Phenyl-3; GL Science, Tokyo, Japan; Φ=6 mm, L=25 cm, 1.6 mL/min), and the eluate was monitored with UV (210 nm) and refractive index monitors. (S)-(+)-2-Methylbutyric acid was purchased from Sigma-Aldrich Corp. (St. Louis, MO, U.S.A.).
Plant MaterialStems of C. cascarilloides Räuschel (Euphorbiaceae) were collected in Kunigami-son, Kunigami-gun, Okinawa, Japan, in July 2004, and a voucher specimen was deposited in the Herbarium of Pharmaceutical Sciences, Graduate School of Biomedical and Health Sciences, Hiroshima University (04-CC-Okinawa-0628).
Extraction and IsolationStems (14.5 kg) of C. cascarilloides were extracted with MeOH (15 L×3) for a week at 25°C. The combined extract was concentrated to 6 L and then partitioned with n-hexane (6 L, n-hexane extract: 92.1 g). The methanolic layer was concentrated and the resulting residue was suspended in 6 L of H2O. The H2O layer was partitioned with 6 L each of CH2Cl2, EtOAc and 1-BuOH to give 39.1 g, 10.5 g and 52.2 g of the respective residues. The residue (39.1 g) of the CH2Cl2-soluble fraction was subjected to silica gel CC (400 g) (Φ=60 mm, L=30 cm) with CHCl3 (5 L), CHCl3–MeOH (15 : 1, 7 L and 12 : 1, 5 L), and MeOH (2 L), fractions of 500 mL being collected. The residue (3.11 g) in fraction 14 was separated by two runs of ODS open CC [H2O–MeOH (1 : 1, 1 L)→(1 : 9, 1 L) and then H2O–MeOH (1 : 9, 250 mL)→MeOH (250 mL)], fractions of 10 g being collected.
The residue (166 mg) in fractions 45–58 obtained on the second run of ODS open CC was subjected to silica gel CC (15 g, Φ=10 mm, L=40 cm) with a linear gradient solvent system from n-hexane (250 mL) to n-hexane–EtOAc (1 : 1, 250 mL), and then with an isocratic solvent system of n-hexane–EtOAc (1 : 1, 250 mL), fractions of 2 mL being collected. The residue (16.4 mg) in fractions 270–290 was purified by HPLC (ODS-3, H2O–MeOH, 1 : 1) and then the residue (3.9 mg) derived from the peak at 31 min was further purified by HPLC (Phenyl-3, H2O–MeOH, 1 : 1) to give 2.2 mg of 2 and 1.1 mg of 3 from the peaks at 23 and 26 min, respectively. From the peak at 40 min on HPLC on ODS-3, 3.4 mg of 1 was obtained. The residue (47.9 mg) in fractions 330–360 was purified by HPLC (ODS-3, H2O–MeOH, 1 : 1) to give a residue (22.6 mg) from the peak at 32 min, which was further purified by HPLC (Phenyl-3, H2O–CH3CN–MeOH, 7 : 2 : 1) to yield 10.8 mg of 4, 8.0 mg of 5 and 1.1 mg of 7 from the peaks at 27, 29 and 37 min, respectively. The residue (12.3 mg) in fractions 361–400 was subjected to HPLC (ODS-3, H2O–MeOH, 1 : 1) to give a further amount of 7 from the peak at 37 min.
The residue (101 mg) in fractions 64–72 obtained on ODS open CC was subjected to silica gel CC (15 g, Φ=10 mm, L=40 cm) with a linear gradient solvent system from n-hexane (250 mL) to n-hexane–EtOAc (1 : 1, 250 mL), and then with an isocratic solvent system of n-hexane–EtOAc (1 : 1, 250 mL), fractions of 2 mL being collected. The residue (12.2 mg) in fractions 152–165 was purified by HPLC (Phenyl-3, H2O–MeOH, 1 : 1) to give 4.1 mg of 6 from the peak at 41 min.
Crotocascarin L (1)Amorphous powder, [α]D24 +6.8 (c=0.22, CHCl3); IR νmax (KBr) cm−1: 3435, 2968, 2929, 1747, 1645, 1443, 1231; UV λmax (MeOH) nm (log ε): 216 (3.97); 1H-NMR (CDCl3. 400 MHz): Table 1; 13C-NMR (CDCl3, 100 MHz): Table 2; CD ∆ε (nm): −15.8 (225), +6.17 (251) (c=2.19×10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 425.1562 [M+Na]+ (Calcd for C22H26O7Na: 425.1570).
Crotocascarin M (2)Colorless needles (MeOH), mp 115–117°C, [α]D24 +44.2 (c=0.15, CHCl3); IR νmax (KBr) cm−1: 3435, 1765, 1634, 1444, 1384; UV λmax (MeOH) nm (log ε): 219 (3.82); 1H-NMR (CDCl3. 400 MHz): Table 1; 13C-NMR (CDCl3, 100 MHz): Table 2; CD ∆ε (nm): −11.7 (223), +4.08 (250) (c=3.92×10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 397.1620 [M+Na]+ (Calcd for C21H26O6Na: 397.1621).
Crotocascarin N (3)Amorphous powder, [α]D26 +2.8 (c=0.45, CHCl3); IR νmax (KBr) cm−1: 3480, 2982, 2962, 2925, 1739, 1455, 1262, 1015; UV λmax (MeOH) nm (log ε): 223 (3.79); 1H-NMR (CDCl3. 400 MHz): Table 1; 13C-NMR (CDCl3, 100 MHz): Table 2; CD ∆ε (nm): −2.30 (224), +1.46 (252) (c=1.32×10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 381.1297 [M+Na]+ (Calcd for C20H22O6Na: 381.1308).
Crotocascarin O (4)Amorphous powder, [α]D25 +103.1 (c=0.72, CHCl3); IR νmax (KBr) cm−1: 3502, 2972, 2931, 1743, 1652, 1458, 1339, 1195, 1151, 929; UV λmax (MeOH) nm (log ε): 218 (3.81); 1H-NMR (CDCl3. 400 MHz): Table 1; 13C-NMR (CDCl3, 100 MHz): Table 2; CD ∆ε (nm): −10.0 (229), +5.31 (254) (c=5.38×10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 501.2103 [M+Na]+ (Calcd for C25H34O9Na: 501.2095).
Crotocascarin P (5)Amorphous powder, [α]D25 +96.9 (c=0.53, CHCl3); IR νmax (KBr) cm−1: 3481, 2972, 2935, 1738, 1653, 1458, 1338, 1197, 1148, 925; UV λmax (MeOH) nm (log ε): 223 (3.88); 1H-NMR (CDCl3. 400 MHz): Table 1; 13C-NMR (CDCl3, 100 MHz): Table 2; CD ∆ε (nm): +3.20 (218), −0.18 (243), +0.30 (260) (c=7.50×10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 485.2147 [M+Na]+ (Calcd for C25H34O8Na: 485.2145).
Crotocascarin Q (6)Amorphous powder, [α]D25 +146.2 (c=0.27, CHCl3); IR νmax (KBr) cm−1: 3469, 2970, 2930, 1739, 1655, 1637, 1460, 1339, 1070, 906; UV λmax (MeOH) nm (log ε): 219 (3.88); 1H-NMR (CDCl3. 400 MHz): Table 1; 13C-NMR (CDCl3, 100 MHz): Table 2; CD ∆ε (nm): +17.2 (224) (c=3.08×10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 467.2037 [M+Na]+ (Calcd for C25H32O7Na: 467.2040).
Neocrotocascarin (7)Amorphous powder, [α]D24 +5.0 (c=0.08, CHCl3); IR νmax (KBr) cm−1: 3434, 2925, 1776, 1744, 1641, 1634, 1460, 1383, 1178, 1140; 1H-NMR (CDCl3. 400 MHz): Table 1; 13C-NMR (CDCl3, 100 MHz): Table 2; HR-ESI-MS (positive-ion mode) m/z: 483.1995 [M+Na]+ (Calcd for C25H32O8Na: 483.1989).
Acetylation of Crotocascarin O (4) and Neocrotocascarin (7)Crotocascarin O (4) (3.7 mg) was dissolved in a mixture of 500 µL each of pyridine and acetic anhydride. After 12 h, the reaction mixture was evaporated to dryness. The resulting residue (4.7 mg) was crystallized from 2-propanol–CHCl3 to give crotocascarin N triacetate (4a) in a crystalline state. Crotocascarin N triacetate (4a): Colorless plates, mp 160–161°C; [α]D25 –27.0 (c=0.20, CHCl3); IR νmax (KBr) cm−1: 2976, 2936, 2878, 1772, 1753, 1750, 1732, 1651, 1459, 1375, 1200, 1146; UV λmax (MeOH) nm (log ε): 226 (3.91); 1H-NMR (CDCl3. 400 MHz) δ: 5.52 (1H, s, H-18a), 5.39 (1H, d, J=5 Hz, H-1), 5.39 (1H, overlapped, H-11), 5.36 (1H, s, H-18b), 4.86 (1H, s, H-5), 3.77 (1H, d, J=12 Hz, H-13), 3.03 (1H, d, J=12 Hz, H-7), 2.80 (1H, dd, J=15, 8 Hz, H-10a), 2.45 (1H, dd, J=15, 4 Hz, H-10b), 2.42 (1H, dd, J=14, 7 Hz, H-3a), 2.42 (1H, overlapped, H-2′), 2.19 (3H, s, CH3CO–), 2.07 (1H, overlapped, H-2), 2.05, 2.03 (3H each, each s, CH3CO– ×2), 1.84 (3H, s, H3-17), 1.82 (1H, dqd, J=14, 7, 7 Hz, H-3′a), 1.59 (1H, dd, J=14, 10 Hz, H-3b), 1.48 (1H, dqd, J=14, 7, 7 Hz, H-3′b), 1.23 (3H, d, J=7 Hz, H3-5′), 1.13 (3H, s, H3-20), 0.96 (3H, t, J=7 Hz, H3-4′), 0.87 (3H, d, J=7 Hz, H3-19); 13C-NMR (CDCl3, 100 MHz) δ: 175.0 (C-1′), 170.1 (C-16), 169.8, 169.4, 167.5 (–COCH3 × 3), 156.0 (C-8), 143.3 (C-12), 132.4 (C-15), 119.8 (C-18), 106.3 (C-9), 74.6 (C-5), 74.3 (C-1), 73.7 (C-6), 71.4 (C-11), 68.5 (C-14), 63.5 (C-4), 43.6 (C-7), 42.5 (C-10), 41.3 (C-2′), 38.2 (C-3), 32.9 (C-2), 31.5 (C-13), 26.1 (C-3′), 23.1 (C-20), 21.8, 21.3, 20.8 (–COCH3 × 3), 16.8 (C-4′), 12.6 (C-19), 11.8 (C-5′), 10.6 (C-17); CD ∆ε (nm): −18.9 (228), +3.71 (255) (c=3.31×10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 627.2418 [M+Na]+ (Calcd for C31H40O12Na: 627.2411).
Neocrotocascarin (7) (0.6 mg) was dissolved in a mixture of 250 µL each of pyridine and acetic anhydride. After 3 h, the reaction mixture was evaporated to dryness. The resulting residue (0.7 mg) was crystallized from n-hexane–CHCl3 to give neocrotocascarin monoacetate (7a) as a crystalline state. Neocrotocascarin monoacetate (7a): colorless rods, mp 284–285°C; 1H-NMR (CDCl3, 400 MHz) δ: 5.79 (1H, s, H-5), 5.61 (1H, d, J=5 Hz, H-1), 5.29 (1H, s, H-18a), 5.27 (1H, s, H-18b), 5.03 (1H, br d, J=8 Hz, H-11), 3.13 (1H, d, J=12 Hz, H-13), 3.11 (1H, d, J=9 Hz, H-8), 2.51 (1H, dd, J=12, 9 Hz, H-7), 2.48 (1H, dd, J=13, 8 Hz, H-10a), 2.43 (1H, overlapped, H-2′), 2.18 (3H, s, CH3CO–), 2.15 (1H, overlapped, H-3a), 2.13 (1H, m, H-2), 2.06 (1H, br d, J=13 Hz, H-10b), 1.75 (3H, s, H3-17), 1.70 (1H, dqd, J=14, 7, 7 Hz, H-3′a), 1.56 (1H, overlapped, H-3b), 1.45 (1H, dqd, J=14, 7, 7 Hz, H-3′b), 1.40 (3H, s, H3-20), 1.17 (3H, d, J=7 Hz, H3-5′), 0.90 (3H, t, J=7 Hz, H3-4′), 0.85 (3H, d, J=7 Hz, H3-19); 13C-NMR (CDCl3, 100 MHz) δ: 177.8 (C-16), 175.8 (C-1′), 170.1 (C-9), 169.3 (CH3CO–), 147.8 (C-12), 114.1 (C-18), 83.7 (C-6), 78.2 (C-11), 74.9 (C-1), 74.8 (C-5), 68.1 (C-14), 65.7 (C-4), 51.0 (C-8), 50.3 (C-7), 46.1 (C-10), 41.0 (C-2′), 40.7 (C-15), 35.2 (C-3), 32.8 (C-2), 30.9 (C-13), 26.7 (C-3′), 24.5 (C-17), 23.5 (C-20), 20.6 (CH3CO–), 16.7 (C-5′), 12.5 (C-19), 11.7 (C-4′); HR-ESI-MS (positive-ion mode) m/z: 525.2097 [M+Na]+ (Calcd for C27H34O9Na: 525.2095).
X-Ray Crystallographic Analysis of Crotocascarin N Triacetate (4a)C34H48O13, M=664.72, crystal size: 0.30×0.20×0.08 mm3, space group: orthorhombic, P212121, T=173 K, a=14.992(5) Å, b=19.874(7) Å, c=11.705(7) Å, V=3488(3) Å3, Z=4, Dc=1.266 Mg/m3, F(000)=1424. The data were measured using a Bruker SMART 1000 CCD diffractometer, using MoKα graphite-monochromated radiation (λ=0.71073 Å) in the range of 7.38<2θ<136.4. Of 3547 reflections collected, 3547 were unique (Rint=0.0000, data/restraints/parameters 3547/0/443). The structure was solved by a direct method using the program SHELXTL-97.10) The refinement and all further calculations were carried out using SHELXTL-97. The absorption correction was carried out utilizing the SADABS routine.11) The H atoms were included at calculated positions and treated as riding atoms using the SHELXTL default parameters. The non-H atoms were refined anisotropically using weighted full-matrix least-squares on F2. Final goodness-of-fit on F2=1.038, R1=0.0400, wR2=0.1036 based on I>2σ(I), and R1=0.0588, wR2=0.1131 based on all data. The largest difference peak and hole were 0.518 and −0.333 eÅ−3, respectively. Supplementary X-ray crystallographic data for 3a (CCDC 1484659) can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223–336–033; or deposit@ccdc.cam.ac.uk).
X-Ray Crystallographic Analysis of Neocrotocascarin Monoacetate (7a)C27H34O9, M=502.54, crystal size: 0.15×0.15×0.15 mm3, space group: orthorhombic, P212121, T=120 K, a=9.9719(12) Å, b=10.8716(13) Å, c=23.261(3) Å, V=2521.8(5) Å3, Z=4, Dc=1.324 Mg/m3, F(000)=1072. The data were measured using a Bruker SMART 1000 CCD diffractometer, using MoKα graphite-monochromated radiation (λ=0.71073 Å) in the range of 3.50<2θ<54.9. Of 25688 reflections collected, 5415 were unique (Rint=0.0201, data/restraints/parameters 5415/0/331). The structure was solved by a direct method using the program SHELXTL-97.10) The refinement and all further calculations were carried out using SHELXTL-97. The absorption correction was carried out utilizing the SADABS routine.11) The H atoms were included at calculated positions and treated as riding atoms using the SHELXTL default parameters. The non-H atoms were refined anisotropically using weighted full-matrix least-squares on F2. Final goodness-of-fit on F2=1.030, R1=0.0286, wR2=0.07257 based on I>2σ(I), and R1=0.0305, wR2=0.0739 based on all data. The largest difference peak and hole were 0.206 and −0.145 eÅ−3, respectively. Supplementary X-ray crystallographic data for 3 (CCDC 1484658) can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223–336–033; or deposit@ccdc.cam.ac.uk).
HPLC Analysis of 2-Methylbutyric Acid in Crotocascarin O (4) and Neocrotocascarin (7)Crotocascarin O (4) (0.5 mg), neocrotocascarin (7) (0.3 mg), and authentic (S)-(+)-2-methylbutyric acid (500 µL) were each dissolved in 1 mL of a 1 : 1 mixture of 10% KOH in H2O and 50% aqueous 1,4-dioxane, and then heated for 3 h at 100°C. The cooled reaction mixtures were acidified with one drop of conc. HCl and then the filtrates were evaporated. The three residues were analyzed by HPLC (column: Inertsil ODS-3, 6×250 mm; solvent: 20% acetonitrile in H2O containing 0.5% phosphoric acid; flow rate: 1.6 mL/min) with a chiral detector (JASCO OR-4090), which revealed a peak of (S)-(+)-2-methylbutyric acid at 16.8 min with a positive optical rotation sign.