Experimental
ChemistryTLC analyses were done using pre-coated silica gel plates. The NMR spectra were recorded on a Bruker AV III-500 spectrometer, the NMR chemical shifts in δ (ppm) were referenced to the solvent peaks of δC 77.0 and δH 7.26 for CDCl3, and chemical shifts were given in δ (ppm) and signals were described as singlet (s), doublet (d), triplet (t), double doublet (dd) and multiplet (m). The high-resolution (HR) electrospray ionization-time of flight (ESI-TOF)-MS was recorded on Agilent 6224 A (TOF) LC/MS. All the solvents used were analytical grade.
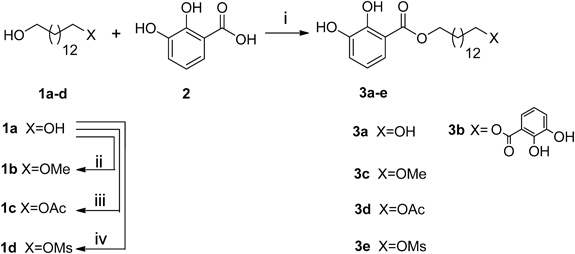
14-Methoxy-1-tetradecanol (1b)Tetradecane-1,14-diol (1a, 690 mg, 3.00 mmol) was dissolved in anhydrous N,N-dimethylformamide (DMF) (20 mL). NaH (180 mg, 4.5 mmol) was added in at 0°C, kept stirring for 30 min. Warmed up to room temperature for another 30 min. MeI (511 mg, 3.60 mmol) was added in at 0°C, kept stirring overnight. The reaction was stopped by adding 1 mL of methanol, the mixture was washed with 1 N HCl solution, water, saturated aqueous solution of NaHCO3 and brine, successively. The organic phase was dried over Na2SO4, filtered, and then concentrated. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 20 : 1) on silica gel to afford 1b (424 mg, 58%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 3.65 (2H, t, J=6.5 Hz), 3.36 (2H, t, J=6.5 Hz), 3.33 (3H, s), 1.57–1.53 (4H, m), 1.32–1.25 (20H, m).
1-Hydroxytetradecyl Acetate (1c)Tetradecane-1,14-diol (1a, 460 mg, 2.00 mmol), Et3N (1.01 g, 10.0 mmol) 4-(dimethylamino)pyridine (DMAP) (12 mg, 0.2 mmol) were dissolved in anhydrous CH2Cl2 (100 mL). Cooled down the mixture to 0°C and Ac2O (225 mg, 2.20 mmol) was added, and kept stirring overnight. The reaction was stopped by adding 1 mL of methanol, the mixture was washed with 1 N HCl solution, saturated aqueous solution of NaHCO3 and brine, successively. The organic phase was dried over Na2SO4, filtered, and then concentrated. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 10 : 1) on silica gel to afford 1c (234 mg, 43%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 4.05 (2H, t, J=7.0 Hz), 3.64 (2H, t, J=6.5 Hz), 2.04 (3H, s), 1.63–1.54 (4H, m), 1.34–1.26 (20H, m).
1-Hydroxytetradecyl Methanesulphonate (1d)Tetradecane-1,14-diol (1a, 2.30 g, 10.00 mmol) was dissolved in anhydrous pyridine (20 mL), DMAP (122 mg, 1.0 mmol), MsCl (0.77 mL, 10.00 mmol) were added into this mixture at 0°C. Then warmed up to room temperature and keep stirring for 6h. The reaction was stopped with 2 mL methanol, the mixture was washed with 1 N HCl solution, saturated aqueous solution of NaHCO3 and brine, successively. The organic phase was dried over Na2SO4, filtered, and then concentrated. The crude mixture was purified by column chromatography (CH2Cl2–MeOH 100 : 1) on silica gel to afford 1d (920 mg, 30%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) 4.22 (2H, t, J=7.0 Hz), 3.64 (2H, t, J=6.5 Hz), 3.00 (3H, s), 1.75 (2H, m), 1.56 (2H, m), 1.40–1.26 (20H, m).
14-Hydroxytetradecyl 2,3-Dihydroxybenzoate (3a) and 1,14-(2,3-Dihydroxybenzoyloxy)tetradecane (3b)Tetradecane-1,14-diol (1a, 230 mg, 1.00 mmol) was dissolved in anhydrous CH2Cl2 (30 mL), DMAP (122 mg, 1.0 mmol), EDC·HCl (1.92 g, 10.0 mmol) and 2,3-dihydroxybenzoic acid (1.54 g, 10.00 mmol) was added into this mixture. Kept stirring overnight at room temperature, and then the mixture was concentrated under vacuum. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 30 : 1) on silica gel to afford 3a (101 mg, 20%) as a colorless powder and 3b (181 mg, 48%) as a colorless powder. 3a: 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.67 (1H, s), 4.34 (2H, t, J=7.0 Hz), 3.64 (2H, t, J=6.5 Hz), 1.77 (2H, m), 1.56 (2H, m), 1.43 (2H, m), 1.35–1.26 (18H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.7, 65.7, 63.1, 32.8, 29.6, 29.5, 29.4, 29.2, 28.5, 25.9, 25.7. HR-ESI-MS m/z: 389.2311 (Calcd for C21H34O5Na [M+Na]+: 389.2298). 3b: 1H-NMR (500 MHz, CDCl3) δ: 10.99 (2H, s), 7.37 (2H, d, J=8.0 Hz), 7.10 (2H, d, J=8.0 Hz), 6.79 (2H, t, J=8.0 Hz), 5.65 (2H, s), 4.34 (4H, t, J=7.0 Hz), 1.79–1.75 (4H, m), 1.45–1.40 (4H, m), 1.37–1.27 (16H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.7, 65.7, 29.6, 29.5, 29.2, 28.5, 25.9. HR-ESI-MS m/z: 525.2459 (Calcd for C28H38O8Na [M+Na]+: 525.2459).
14-Methoxytetradecyl 2,3-Dihydroxybenzoate (3c)Compound 1b (244 mg, 1.00 mmol) was dissolved in anhydrous CH2Cl2 (20 mL), DMAP (12 mg, 0.1 mmol), EDC·HCl (575 mg, 3.0 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol) were added into this mixture. Kept stirring overnight at room temperature, and then the mixture was concentrated under vacuum. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 20 : 1) on silica gel to afford 3c (181 mg, 48%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 10.99 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.79 (1H, t, J=8.0 Hz), 5.68 (1H, s), 4.34 (2H, t, J=7.0 Hz), 3.36 (2H, t, J=6.5 Hz), 3.33 (3H, s), 1.78 (2H, m), 1.59–1.55 (2H, m), 1.43–1.26 (20H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.7, 73.0, 65.7, 58.5, 29.6, 29.5, 29.2, 28.5, 26.1, 25.9. HR-ESI-MS m/z: 403.2451 (Calcd for C22H36O5Na [M+Na]+: 403.2455).
14-Acetoxytetradecyl 2,3-Dihydroxybenzoate (3d)The synthesis and purification methods were the same as for 3c, condensing 1c (100 mg, 0.37 mmol) with 2,3-dihydroxybenzoic acid (113 mg, 0.73 mmol). The pure 3d was obtained as a colorless powder (72 mg, 48%). 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.65 (1H, s), 4.34 (2H, t, J=6.5 Hz), 4.05 (2H, t, J=7.0 Hz), 2.05 (3H, s), 1.78 (2H, m), 1.60 (2H, m), 1.45–1.26 (20H, m). 13C-NMR (125 MHz, CDCl3) δ: 171.3, 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.7, 65.7, 64.7, 29.6, 29.5, 29.3, 29.2, 28.6, 28.5, 25.9, 21.0. HR-ESI-MS m/z: 431.2372 (Calcd for C23H36O6Na [M+Na]+: 431.2404).
14-(Methylsulphonyloxy)tetradecyl 2,3-Dihydroxybenzoate (3e)Compound 1d (920 mg, 2.98 mmol) was dissolved in anhydrous CH2Cl2 (20 mL), DMAP (37 mg, 0.3 mmol), EDC·HCl (1.14 g, 6.0 mmol) were added into this mixture. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 5 : 1) on silica gel to afford 3e (540 mg, 41%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) 10.99 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.64 (1H, s), 4.35 (2H, t, J=6.5 Hz), 4.22 (2H, t, J=6.5 Hz), 3.00 (3H, s), 1.79–1.73 (4H, m), 1.44–1.26 (20H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.7, 70.2, 65.7, 37.4, 29.6, 29.5, 29.4, 29.2, 29.1, 29.0, 28.5, 25.9, 25.4. HR-ESI-MS m/z: 467.2081 (Calcd for C22H36O7SNa [M+Na]+: 467.2074).
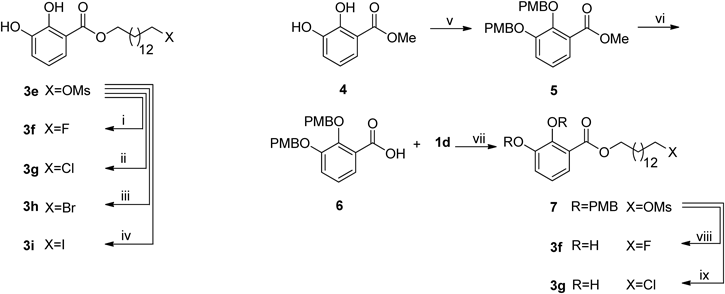
Methyl 2,3-Bis(4-methoxybenzyloxy)benzoate (5)Methyl 2,3-dihydroxybenzoate (4, 1.00 g, 5.94 mmol) was dissolved in anhydrous tetrahydrofuran (THF) (50 mL), K2CO3 (4.92 g, 35.6 mmol), tetrabutylammonium iodide (TBAI) (0.22 g, 0.6 mmol), and PMBCl (2.42 mL, 17.82 mmol) were added into the solution. The mixture was refluxed for 24 h, and then filtered. The filtrate was concentrated, and purified by column chromatography (petroleum ether–EtOAc 50 : 1) to afford 5 (1.70 g, 70%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 7.40–7.35 (5H, m), 7.14 (1H, dd, J=8.5, 1.5 Hz), 7.07 (1H, t, J=8.0 Hz), 6.92 (2H, d, J=8.5 Hz), 6.85 (2H, d, J=8.5 Hz), 5.06 (2H, s), 5.04 (2H, s), 3.87 (3H, s), 3.82 (3H, s), 3.80 (3H, s).
2,3-Bis(4-methoxybenzyloxy)benzoic Acid (6)A solution of 5 (0.94 g, 1.83 mmol) in dioxane (9 mL) and 2 N NaOH (4.6 mL) was stirred for 24 h at room temperature. The reaction mixture was concentrated. The residue was stirred with H2O (5 mL) and then acidified to pH 2 with 2 N HCl. The colorless powder was filtered, washed with hexane, and recrystallized from EtOAc–hexane to yield 500 mg (69%) of 6 as a colorless crystal. 1H-NMR (500 MHz, CDCl3) δ: 7.72 (1H, dd, J=8.0, 2.0 Hz), 7.41 (2H, d, J=9.0 Hz), 7.27–7.22 (3H, m), 7.18 (1H, t, J=8.0 Hz), 6.97–6.95 (2H, m), 6.84–6.82 (2H, m), 5.20 (2H, s), 5.12 (2H, s), 3.85 (3H, s), 3.80 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 165.2, 160.3, 159.8, 151.3, 147.0, 131.1, 129.6, 127.9, 126.7, 124.9, 124.3, 122.9, 119.0, 114.1, 76.8, 71.3, 55.3, 55.2.
14-(Methylsulphonyloxy)tetradecyl 2,3-Dihydroxybenzoate (7)Compound 1d (270 mg, 0.88 mmol) was dissolved in anhydrous CH2Cl2 (20 mL), DMAP (11 mg, 0.1 mmol) and EDC·HCl (503 mg, 2.6 mmol), and 6 (686 mg, 1.75 mmol) were added into this mixture. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 5 : 1) on silica gel to yield 7 (259 mg, 43%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 7.37–7.32 (5H, m), 7.12 (1H, d, J=8.0 Hz), 7.06 (1H, t, J=8.0 Hz), 6.91 (2H, m), 6.82 (2H, m), 5.05 (2H, s), 5.02 (2H, s), 4.26 (2H, t, J=7.0 Hz), 4.22 (2H, t, J=7.0 Hz), 3.83 (3H, s), 3.80 (3H, s), 2.99 (3H, s), 1.77–1.67 (4H, m), 1.41–1.35 (4H, m), 1.31–1.23 (16H, m). 13C-NMR (125 MHz, CDCl3) δ: 166.5, 159.5, 159.3, 152.8, 148.2, 130.2, 129.8, 129.3, 128.7, 127.3, 123.8, 122.6, 117.9, 113.9, 113.6, 77.2, 75.2, 71.1, 70.2, 65.3, 55.3, 55.2, 37.3, 29.6, 29.5, 29.4, 29.3, 29.1, 29.0, 28.7, 26.0, 25.4.
14-Fluorotetradecyl 2,3-Dihydroxybenzoate (3f)Compound 7 (10 mg, 0.015 mmol) was dissolved in 2 mL anhydrous acetonitrile, KF (1 mg, 0.2 mmol), K222 (20 mg, 0.1 mmol) were added, the reaction mixture was refluxed for 15 min. Solvent was evaporated under vacuum to give a crude product. The crude mixture was dissolved in 1 mL CH2Cl2, then 1 mL TFA was added in with stirring at room temperature, kept stirring for 10 min. Then the mixture was diluted with 10 mL distilled water, and subjected to C18 column, washed with 2 mL distilled water, 2 mL EtOH (50%) to yield 3f (4.1 mg, 76%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.63 (1H, s), 4.44 (2H, dt, J=47.0, 6.5 Hz), 4.34 (2H, t, J=6.5 Hz), 1.80–1.63 (4H, m), 1.45–1.27 (20H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.9, 120.5, 119.6, 119.1, 112.7, 84.3 (d, J=162.9 Hz), 65.7, 30.4, 30.3, 29.6, 29.5, 29.2, 28.5, 25.9, 25.1. HR-ESI-MS m/z: 391.2265 (Calcd for C21H33O4FNa [M+Na]+: 391.2255).
14-Chlorotetradecyl 2,3-Dihydroxybenzoate (3g)The synthesis and purification methods were the same as for 3f, starting from compound 7 (68 mg, 0.10 mmol), K222 (75 mg, 0.2 mmol) and KCl (37 mg, 0.5 mmol). The pure 3g was obtained as a colorless powder (30 mg, 79%). 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.64 (1H, s), 4.35 (2H, t, J=6.5 Hz), 3.53 (2H, t, J=6.5 Hz), 1.81–1.74 (4H, m), 1.45–1.41 (4H, m), 1.37–1.27 (16H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.7, 65.7, 45.2, 32.6, 29.6, 29.5, 29.4, 29.2, 28.9, 28.5, 26.9, 25.9. HR-ESI-MS m/z: 407.1956 (Calcd for C21H33O4ClNa [M+Na]+: 407.1960).
14-Bromotetradecyl 2,3-Dihydroxybenzoate (3h)Compound 3e (44 mg, 0.10 mmol) was dissolved in 2 mL anhydrous acetonitrile, KBr (36 mg, 0.3 mmol), K222 (38 mg, 0.1 mmol) were added, the reaction mixture was refluxed for 30 min. Solvent was evaporated under vacuum to give the crude product which was purified by column chromatography (petroleum ether–EtOAc 5 : 1) on silica gel to afford 3h (38 mg, 88%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 10.99 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.64 (1H, s), 4.34 (2H, t, J=6.5 Hz), 3.41 (2H, t, J=6.5 Hz), 1.87–1.75 (4H, m), 1.45–1.27 (20H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.6, 119.1, 112.7, 65.7, 34.0, 32.8, 29.6, 29.5, 29.4, 29.2, 28.8, 28.5, 28.2, 25.9. HR-ESI-MS m/z: 451.1457 (Calcd for C21H33O479BrNa [M+Na]+: 451.1454).
14-Iodotetradecyl 2,3-Dihydroxybenzoate (3i)Compound 3e (44 mg, 0.10 mmol) was dissolved in anhydrous acetonitrile, KI (33 mg, 0.2 mmol), K222 (38 mg, 0.1 mmol) were added, the reaction mixture was refluxed for 30 min. Solvent was evaporated to give the crude which was purified by column chromatography (hexane–EtOAc 30 : 1) on silica gel to afford 3i (44 mg, 92%) as a colorless powder. 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.66 (1H, s), 4.34 (2H, t, J=6.5 Hz), 3.19 (2H, t, J=7.0 Hz), 1.85–1.75 (4H, m), 1.46–1.26 (20H, m). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.6, 119.1, 112.7, 65.7, 33.6, 30.5, 29.6, 29.5, 29.4, 29.2, 28.5, 25.9, 7.3. HR-ESI-MS m/z: 499.1330 (Calcd for C21H33O4INa [M+Na]+: 499.1316).
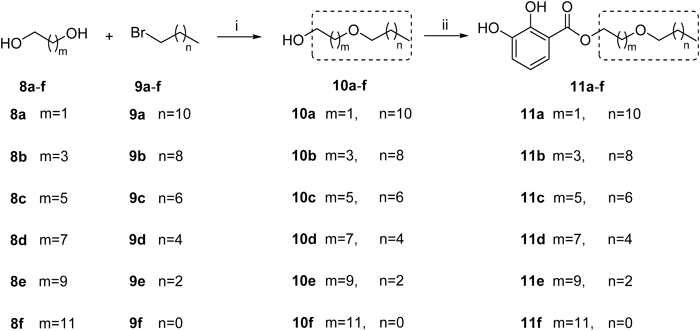
2-(Dodecyloxy)ethanol (10a)Ethanediol (1.1 mL, 20.00 mmol) was dissolved in 200 mL anhydrous DMF, NaH (480 mg, 12.0 mmol) was added in at 0°C. The mixture was warmed up to room temperature and stirred for 30 min. Then 1-bromododecane (2.4 mL, 10.00 mmol), TBAI (369 mg, 1.0 mmol) were added in at 0°C, kept stirring overnight. The reaction mixture was diluted with 20 mL methanol, then washed with 1 N HCl solution, water, saturated aqueous solution of NaHCO3 and brine, successively. The organic phase was dried over Na2SO4, filtered, and then concentrated. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 30 : 1) on silica gel to afford 10a as a colorless oil (1.12 g, 49%). 1H-NMR (500 MHz, CDCl3) δ: 3.73 (2H, t, J=4.5 Hz), 3.53 (2H, t, J=4.5 Hz), 3.47 (2H, t, J=6.5 Hz), 1.59 (2H, m), 1.29–1.26 (18H, m), 0.88 (3H, t, J=6.5 Hz).
4-(Decyloxy)butan-1-ol (10b)The synthesis and purification methods were the same as for 10a. The pure 10b was obtained as a colorless oil (1.01 g, 44%). 1H-NMR (500 MHz, CDCl3) δ: 3.64 (2H, t, J=5.5 Hz), 3.45 (2H, t, J=5.5 Hz), 3.42 (2H, t, J=6.5 Hz), 1.70–1.65 (4H, m), 1.56 (2H, m), 1.30–1.25 (14H, m), 0.87 (3H, t, J=7.0 Hz).
6-(Octyloxy)hexan-1-ol (10c)The synthesis and purification methods were the same as for 10a. The pure 10c was obtained as a colorless oil (1.09 g, 47%). 1H-NMR (500 MHz, CDCl3) δ: 3.60 (2H, t, J=6.5 Hz), 3.39–3.35 (4H, m), 1.58–1.51 (6H, m), 1.35–1.25 (14H, m), 0.85 (3H, t, J=6.5 Hz).
8-(Hexyloxy)octan-1-ol (10d)The synthesis and purification methods were the same as for 10a. The pure 10d was obtained as a colorless oil (0.91 g, 39%). 1H-NMR (500 MHz, CDCl3) δ: 3.63 (2H, t, J=6.5 Hz), 3.39 (4H, t, J=6.5 Hz), 1.57–1.53 (6H, m), 1.36–1.25 (14H, m), 0.88 (3H, t, J=6.5 Hz).
10-(Butoxy)decan-1-ol (10e)The synthesis and purification methods were the same as for 10a. The pure 10e was obtained as a colorless oil (1.11 g, 48%). 1H-NMR (500 MHz, CDCl3) δ: 3.64 (2H, t, J=6.5 Hz), 3.41–3.37 (4H, m), 1.58–1.52 (6H, m), 1.40–1.28 (14H, m), 0.92 (3H, t, J=7.0 Hz).
12-(Ethoxy)dodecan-1-ol (10f)The synthesis and purification methods were the same as for 10a. The pure 10f was obtained as a colorless oil (0.99 g, 43%). 1H-NMR (500 MHz, CDCl3) δ: 3.63 (2H, t, J=6.5 Hz), 3.46 (2H, m, J=7.0 Hz), 3.40 (2H, t, J=7.0 Hz), 1.58–1.52 (4H, m), 1.33–1.26 (16H, m), 1.19 (3H, t, J=7.0 Hz).
2-(Dodecyloxy)ethyl 2,3-Dihydroxybenzoate (11a)The synthesis and purification methods were the same as for 3a, condensing from 10a (230 mg, 1.00 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 11a was obtained as a colorless powder (181 mg, 49%). 1H-NMR (500 MHz, CDCl3) δ: 10.83 (1H, s), 7.41 (1H, d, J=8.0 Hz), 7.11 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.64 (1H, s), 4.49 (2H, t, J=5.0 Hz), 3.76 (2H, t, J=5.0 Hz), 3.51 (2H, t, J=6.5 Hz), 1.58 (2H, m), 1.34–1.25 (18H, m), 0.88 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 170.2, 148.8, 145.0, 120.8, 119.8, 119.2, 112.5, 71.6, 68.2, 64.6, 31.9, 29.7, 29.6, 29.4, 29.3, 26.1, 22.7, 14.1. HR-ESI-MS m/z: 389.2291 (Calcd for C21H34O5Na [M+Na]+: 389.2298).
4-(Decyloxy)butyl 2,3-Dihydroxybenzoate (11b)The synthesis and purification methods were the same as for 3a, condensing from 10b (230 mg, 1.00 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 11b was obtained as a colorless powder (180 mg, 47%). 1H-NMR (500 MHz, CDCl3) δ: 10.97 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.79 (1H, t, J=8.0 Hz), 5.64 (1H, s), 4.38 (2H, t, J=6.5 Hz), 3.47 (2H, t, J=6.0 Hz), 3.41 (2H, t, J=6.5 Hz), 1.88 (2H, m), 1.73 (2H, m), 1.56 (2H, m), 1.34–1.23 (14H, m), 0.88 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.6, 71.1, 70.1, 65.5, 31.9, 29.7, 29.6, 29.5, 29.3, 26.3, 26.2, 25.5, 22.7, 14.1. HR-ESI-MS m/z: 389.2311 (Calcd for C21H34O5Na [M+Na]+: 389.2298).
6-(Octyloxy)hexyl 2,3-Dihydroxybenzoate (11c)The synthesis and purification methods were the same as for 3a, condensing from 10c (230 mg, 1.00 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 11c was obtained as a colorless solid (170 mg, 44%). 1H-NMR (500 MHz, CDCl3) δ: 10.99 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.79 (1H, t, J=8.0 Hz), 5.64 (1H, s), 4.35 (2H, t, J=6.5 Hz), 3.42–3.38 (4H, m), 1.79 (2H, m), 1.63–1.27 (18H, m), 0.88 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.7, 71.0, 70.7, 65.6, 31.8, 29.8, 29.6, 29.5, 29.3, 28.5, 26.2, 25.9, 25.8, 22.6, 14.1. HR-ESI-MS m/z: 389.2316 (Calcd for C21H34O5Na [M+Na]+: 389.2298).
8-(Hexyloxy)octyl 2,3-Dihydroxybenzoate (11d)The synthesis and purification methods were the same as for 3a, condensing from 10d (230 mg, 1.00 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 11d was obtained as a colorless powder (178 mg, 48%). 1H-NMR (500 MHz, CDCl3) δ: 10.99 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.79 (1H, t, J=8.0 Hz), 5.67 (1H, s), 4.34 (2H, t, J=6.5 Hz), 3.40–3.38 (4H, m), 1.77 (2H, m), 1.59–1.53 (4H, m), 1.45–1.27 (14H, m), 0.88 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.3, 120.5, 120.0, 119.1, 112.7, 71.0, 70.9, 65.7, 31.7, 29.7, 29.3, 29.2, 28.5, 26.1, 25.9, 22.6, 14.0. HR-ESI-MS m/z: 389.2324 (Calcd for C21H34O5Na [M+Na]+: 389.2298).
10-(Butoxy)decyl 2,3-Dihydroxybenzoate (11e)The synthesis and purification methods were the same as for 3a, condensing from 10e (230 mg, 1.00 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 11e was obtained as a colorless powder (190 mg, 52%). 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.67 (1H, s), 4.34 (2H, t, J=6.5 Hz), 3.41–3.78 (4H, m), 1.77 (2H, m), 1.59–1.52 (4H, m), 1.45–1.30 (14H, m), 0.92 (3H, t, J=7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.6, 119.1, 112.7, 70.9, 70.6, 65.7, 31.9, 29.8, 29.5, 29.4, 29.2, 28.5, 26.2, 25.9, 19.4, 13.9. HR-ESI-MS m/z: 389.2293 (Calcd for C21H34O5Na [M+Na]+: 389.2298).
12-(Ethoxy)dodecyl 2,3-Dihydroxybenzoate (11f)The synthesis and purification methods were the same as for 3a, condensing from 10f (230 mg, 1.00 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 11f was obtained as a colorless powder (197 mg, 55%). 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.66 (1H, s), 4.34 (2H, t, J=6.5 Hz), 3.47 (2H, m), 3.40 (2H, t, J=6.5 Hz), 1.77 (2H, m), 1.57 (2H, m), 1.43 (2H, m), 1.34–1.27 (14H, m), 1.20 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 170.4, 148.9, 145.0, 120.5, 119.6, 119.1, 112.7, 70.8, 66.0, 65.7, 29.8, 29.6, 29.5, 29.2, 28.5, 26.2, 25.9, 15.2. HR-ESI-MS m/z: 389.2323 (Calcd for C21H34O5Na [M+Na]+: 389.2298).
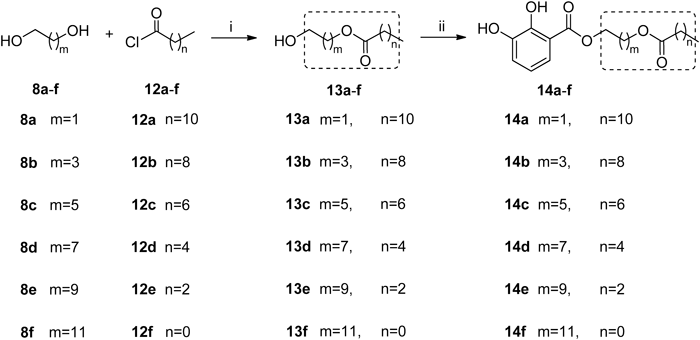
2-Hydroxyethyl Dodecanoate (13a)Ethanediol (1.1 mL, 20.00 mmol), Et3N (5.10 g, 50.0 mmol) and DMAP (122 mg, 1.0 mmol) were dissolved in 200 mL anhydrous CH2Cl2. The C11H23COCl (2.18 g, 10.00 mmol) was added dropwise at 0°C. The mixture was warmed up to room temperature and kept stirring overnight then washed with 1 N HCl solution, water, saturated aqueous solution of NaHCO3 and brine, successively. The organic phase was dried over Na2SO4, filtered, and then concentrated. The crude mixture was purified by column chromatography (petroleum ether–EtOAc 30 : 1) on silica gel to afford 13a as a colorless oil (1.51 g, 62%). 1H-NMR (500 MHz, CDCl3) δ: 4.20 (2H, m), 3.82 (2H, m), 2.34 (2H, t, J=7.5 Hz), 1.62 (2H, m), 1.32–1.25 (16H, m), 0.87 (3H, t, J=6.5 Hz).
4-Hydroxybutyl Decanoate (13b)The synthesis and purification methods were the same as for 13a. The pure 13b was obtained as a colorless oil (1.05 g, 43%). 1H-NMR (500 MHz, CDCl3) δ: 4.10 (2H, t, J=6.5 Hz), 3.68 (2H, t, J=6.5 Hz), 2.29 (2H, t, J=7.5 Hz), 1.72–1.62 (6H, m), 1.30–1.26 (12H, m), 0.87 (3H, t, J=7.0 Hz).
6-Hydroxyhexyl Octanoate (13c)The synthesis and purification methods were the same as for 13a. The pure 13c was obtained as a colorless oil (1.00 g, 41%). 1H-NMR (500 MHz, CDCl3) δ: 4.05 (2H, t, J=7.0 Hz), 3.64 (2H, t, J=7.0 Hz), 2.28 (2H, t, J=7.5 Hz), 1.63–1.55 (6H, m), 1.35–1.29 (12H, m), 0.89 (3H, t, J=7.0 Hz).
8-Hydroxyoctyl Hexanoate (13d)The synthesis and purification methods were the same as for 13a. The pure 13d was obtained as a colorless oil (1.30 g, 53%). 1H-NMR (500 MHz, CDCl3) δ: 4.05 (2H, t, J=7.0 Hz), 3.64 (2H, t, J=7.0 Hz), 2.28 (2H, t, J=7.5 Hz), 1.63–1.55 (6H, m), 1.35–1.29 (12H, m), 0.89 (3H, t, J=7.0 Hz).
10-Hydroxydecyl Butyrate (13e)The synthesis and purification methods were the same as for 13a. The pure 13e was obtained as a colorless oil (1.01 g, 41%). 1H-NMR (500 MHz, CDCl3) δ: 4.05 (2H, t, J=6.5 Hz), 3.63 (2H, t, J=6.5 Hz), 2.27 (2H, t, J=7.5 Hz), 1.67–1.54 (6H, m), 1.38–1.24 (12H, m), 0.94 (3H, t, J=7.5 Hz).
12-Hydroxydodecyl Acetate (13f)The synthesis and purification methods were the same as for 13a. The pure 13f was obtained as a colorless oil (1.10 g, 45%). 1H-NMR (500 MHz, CDCl3) δ: 4.04 (2H, t, J=6.5 Hz), 3.63 (2H, m), 2.04 (3H, s), 1.64–1.53 (4H, m), 1.34–1.26 (16H, m).
2-(Dodecanoyloxy)ethyl 2,3-Dihydroxybenzoate (14a)The synthesis and purification methods were the same as for 3a, condensing from 13a (230 mg, 0.94 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 14a was obtained as a colorless powder (190 mg, 53%). 1H-NMR (500 MHz, CDCl3) δ: 10.75 (1H, s), 7.38 (1H, d, J=8.0 Hz), 7.12 (1H, d, J=8.0 Hz), 6.81 (1H, t, J=8.0 Hz), 4.55 (2H, m), 4.43 (2H, m), 2.34 (2H, t, J=7.5 Hz), 1.61 (2H, m), 1.29–1.24 (16H, m), 0.88 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 173.6, 170.0, 148.9, 145.0, 120.7, 120.0, 119.3, 112.1, 63.2, 61.5, 34.1, 31.9, 29.6, 29.4, 29.3, 29.2, 29.1, 24.9, 22.7, 14.1. HR-ESI-MS m/z: 403.2089 (Calcd for C21H32O6Na [M+Na]+: 403.2091).
4-(Decanoyloxy)butyl 2,3-Dihydroxybenzoate (14b)The synthesis and purification methods were the same as for 3a, condensing from 13b (230 mg, 0.94 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 14b was obtained as a colorless powder (180 mg, 50%). 1H-NMR (500 MHz, CDCl3) δ: 10.93 (1H, s), 7.36 (1H, d, J=8.0 Hz), 7.11 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.64 (1H, s), 4.39 (2H, t, J=6.5 Hz), 4.14 (2H, t, J=6.5 Hz), 2.30 (2H, t, J=7.5 Hz), 1.90–1.77 (4H, m), 1.62 (2H, m), 1.32–1.26 (12H, m), 0.87 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 173.9, 170.3, 148.9, 145.1, 120.5, 119.8, 119.1, 112.4, 65.0, 63.5, 34.3, 31.8, 29.4, 29.2, 29.1, 25.3, 25.0, 22.6, 14.1. HR-ESI-MS m/z: 403.2128 (Calcd for C21H32O6Na [M+Na]+: 403.2091).
6-(Octanoyloxy)hexyl 2,3-Dihydroxybenzoate (14c)The synthesis and purification methods were the same as for 3a, condensing from 13c (230 mg, 0.94 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 14c was obtained as a colorless powder (199 mg, 56%). 1H-NMR (500 MHz, CDCl3) δ: 10.97 (1H, s), 7.36 (1H, d, J=8.0 Hz), 7.11 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.65 (1H, s), 4.35 (2H, t, J=6.5 Hz), 4.08 (2H, t, J=6.5 Hz), 2.29 (2H, t, J=7.0 Hz), 1.79 (2H, m), 1.68–1.57 (4H, m), 1.50–1.41 (4H, m), 1.30–1.27 (8H, m), 0.87 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 174.0, 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.6, 65.5, 64.1, 34.4, 31.7, 29.1, 28.9, 28.5, 28.4, 25.6, 25.0, 22.6, 14.0. HR-ESI-MS m/z: 403.2129 (Calcd for C21H32O6Na [M+Na]+: 403.2091).
8-(Hexanoyloxy)octyl 2,3-Dihydroxybenzoate (14d)The synthesis and purification methods were the same as for 3a, condensing from 13d (230 mg, 0.94 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 14d was obtained as a colorless powder (177 mg, 49%). 1H-NMR (500 MHz, CDCl3) δ: 10.99 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.80 (1H, t, J=8.0 Hz), 5.65 (1H, s), 4.34 (2H, t, J=7.0 Hz), 4.06 (2H, t, J=7.0 Hz), 2.29 (2H, t, J=7.5 Hz), 1.78 (2H, m), 1.64–1.61 (4H, m), 1.44–1.29 (12H, m), 0.89 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 174.0, 170.4, 148.9, 145.0, 120.5, 119.7, 119.1, 112.6, 65.6, 64.3, 34.4, 31.3, 29.1, 28.6, 28.5, 25.8, 24.7, 22.3, 13.9. HR-ESI-MS m/z: 403.2118 (Calcd for C21H32O6Na [M+Na]+: 403.2091).
10-(Butyryloxy)decyl 2,3-Dihydroxybenzoate (14e)The synthesis and purification methods were the same as for 3a, condensing from 13e (230 mg, 0.94 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 14e was obtained as a colorless powder (190 mg, 53%). 1H-NMR (500 MHz, CDCl3) δ: 10.98 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.79 (1H, t, J=8.0 Hz), 5.69 (1H, s), 4.34 (2H, t, J=6.5 Hz), 4.06 (2H, t, J=6.5 Hz), 2.27 (2H, t, J=7.5 Hz), 1.78 (2H, m), 1.67–1.26 (16H, m), 0.95 (3H, t, J=7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ: 173.8, 170.4, 148.9, 145.1, 120.5, 119.7, 119.1, 112.7, 65.7, 64.3, 36.3, 29.4, 29.2, 28.6, 28.5, 25.9, 18.5, 13.7. HR-ESI-MS m/z: 403.2092 (Calcd for C21H32O6Na [M+Na]+: 403.2091).
12-(Acetoxy)dodecyl 2,3-Dihydroxybenzoate (14f)The synthesis and purification methods were the same as for 3a, condensing from 13f (230 mg, 0.94 mmol) and 2,3-dihydroxybenzoic acid (308 mg, 2.00 mmol). The pure 14f was obtained as a colorless powder (203 mg, 57%). 1H-NMR (500 MHz, CDCl3) δ: 11.00 (1H, s), 7.37 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.79 (1H, t, J=8.0 Hz), 5.78 (1H, s), 4.34 (2H, t, J=6.5 Hz), 4.05 (2H, t, J=6.5 Hz), 2.05 (3H, s), 1.77 (2H, m), 1.61 (2H, m), 1.45–1.27 (16H, m). 13C-NMR (125 MHz, CDCl3) δ: 171.2, 170.4, 148.9, 145.0, 120.5, 119.6, 119.1, 112.7, 65.7, 64.7, 29.5, 29.2, 28.6, 28.5, 25.9, 21.0. HR-ESI-MS m/z: 403.2094 (Calcd for C21H32O6Na [M+Na]+: 403.2091).
S-Hexyl 2,3-Dihydroxybenzothioate (15a)To a solution of compound 6 (79 mg, 0.20 mmol), triethylamine (202 mg, 2.0 mmol) in dry CH2Cl2 (5 mL) at 0°C was added pivaloyl chloride (24 mg, 0.20 mmol). The mixture was warmed to room temperature and was stirred until compound 6 disappeared (detected by TLC). Then 1-hexanethiol (47 mg, 0.40 mmol) and 4-dimethylaminopyridine (2.4 mg, 0.02 mmol) were added into the mixture at 0°C, warmed up to room temperature, and kept stirring for 12 h. Finally, trifluoroacetic acid (50% in water, 5 mL) was added into the mixture, and kept stirring for 12 h. The mixture was extracted with CH2Cl2, and washed with water, saturated aqueous solution of NaHCO3 and brine, successively. The organic phase was dried over Na2SO4, filtered, and then concentrated. The crude mixture was purified by column chromatography (n-hexane–EtOAc 300 : 1) on silica gel to afford 15a as a colorless oil (27 mg, 53%). 1H-NMR (500 MHz, CDCl3) δ: 11.26 (1H, s), 7.42 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.81 (1H, t, J=8.0 Hz), 5.69 (1H, s), 3.07 (2H, t, J=7.0 Hz), 1.68 (2H, m), 1.44 (2H, m), 1.33–1.31 (4H, m), 0.90 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 197.9, 146.6, 145.3, 120.0, 119.7, 119.5, 119.4, 31.8, 29.3, 29.1, 28.9, 22.6, 14.1. HR-ESI-MS m/z: 255.1063 (Calcd for C13H19O3S [M+H]+: 255.1049).
S-Octyl 2,3-Dihydroxybenzothioate (15b)The synthesis and purification methods were the same as for 15a using 1-octanethiol (29 mg, 0.20 mmol) as corresponding thiol, the pure 15b was obtained as a colorless oil (30 mg, 53%). 1H-NMR (500 MHz, CDCl3) δ: 11.26 (1H, s), 7.42 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.81 (1H, t, J=8.0 Hz), 5.69 (1H, s), 3.07 (2H, t, J=7.0 Hz), 1.68 (2H, m), 1.41–1.40 (2H, m), 1.34–1.28 (8H, m), 0.89 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 197.9, 146.6, 145.3, 120.0, 119.7, 119.5, 119.4, 31.8, 29.3, 29.1, 28.9, 22.6, 14.1. HR-ESI-MS m/z: 283.1369 (Calcd for C15H23O3S [M+H]+: 283.1362).
S-Decyl 2,3-Dihydroxybenzothioate (15c)The synthesis and purification methods were the same as for 15a using 1-decanethiol (35 mg, 0.20 mmol) as corresponding thiol, the pure 15c was obtained as a colorless oil (35 mg, 56%). 1H-NMR (500 MHz, CDCl3) δ: 11.26 (1H, s), 7.42 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.81 (1H, t, J=8.0 Hz), 5.69 (1H, s), 3.07 (2H, t, J=7.5 Hz), 1.68 (2H, m), 1.42 (2H, m), 1.34–1.27 (12H, m), 0.88 (3H, t, J=7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 197.9, 146.6, 145.3, 120.0, 119.7, 119.5, 119.4, 31.9, 29.5, 29.3, 29.1, 28.9, 22.7, 14.1. HR-ESI-MS m/z: 311.1687 (Calcd for C17H27O3S [M+H]+: 311.1675).
S-Dodecyl 2,3-Dihydroxybenzothioate (15d)The synthesis and purification methods were the same as for 15a using 1-dodecanethiol (40 mg, 0.20 mmol) as corresponding thiol, the pure 15d was obtained as a colorless oil (39 mg, 58%). 1H-NMR (500 MHz, CDCl3) δ: 11.23 (1H, s), 7.42 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.81 (1H, t, J=8.0 Hz), 5.67 (1H, s), 3.07 (2H, t, J=7.5 Hz), 1.68 (2H, m), 1.42 (2H, m), 1.34–1.26 (16H, m), 0.88 (3H, t, J=6.5 Hz). 13C-NMR (125 MHz, CDCl3) δ: 197.880, 146.642, 145.300, 120.001, 119.689, 119.492, 119.350, 31.904, 29.620, 29.557, 29.462, 29.330, 29.287, 29.104, 28.891, 22.676, 14.098. HR-ESI-MS m/z: 339.1989 (Calcd for C19H31O3S [M+H]+: 339.1988).
S-Tetradecyl 2,3-Dihydroxybenzothioate (15e)The synthesis and purification methods were the same as for 15a using 1-tetradecanethiol (46 mg, 0.20 mmol) as corresponding thiol, the pure 15e was obtained as a colorless powder (37 mg, 50%). 1H-NMR (500 MHz, CDCl3) δ: 11.27 (1H, s), 7.42 (1H, d, J=8.0 Hz), 7.10 (1H, d, J=8.0 Hz), 6.81 (1H, t, J=8.0 Hz), 5.67 (1H, s), 3.07 (2H, t, J=6.5 Hz), 1.68 (2H, m), 1.42 (2H, m), 1.34–1.26 (20H, m), 0.88 (3H, t, J=6.5 Hz). 13C-NMR (125 MHz, CDCl3) δ: 197.881, 146.643, 145.299, 120.000, 119.690, 119.490, 119.350, 31.914, 29.643, 29.559, 29.463, 29.346, 29.288, 29.104, 28.883, 22.680, 14.101. HR-ESI-MS m/z: 367.2300 (Calcd for C21H35O3S [M+H]+: 367.2301).
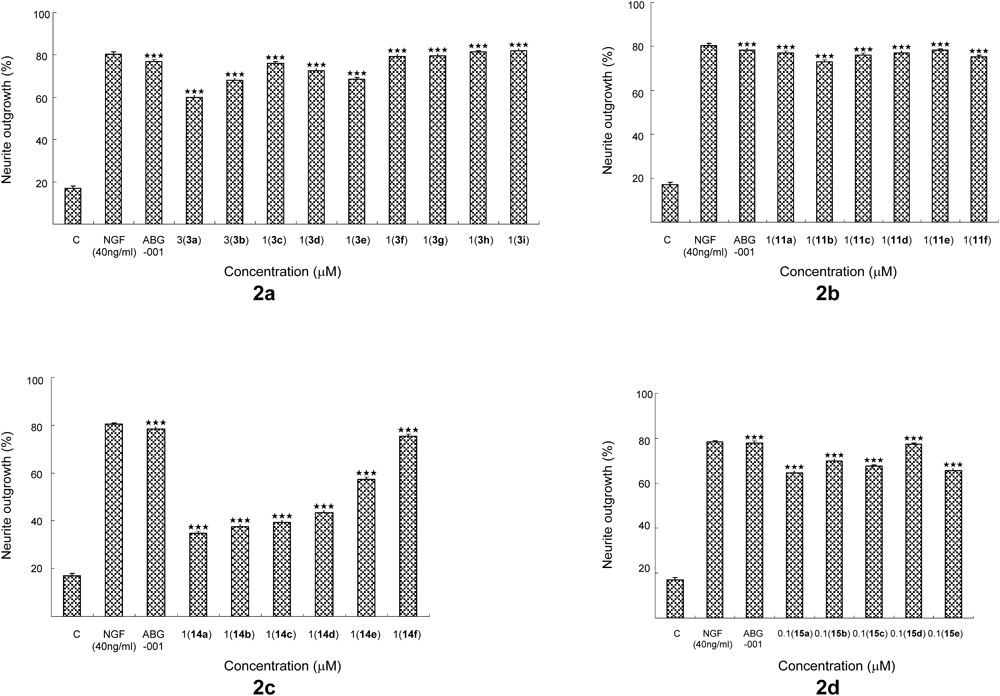
BioassayBiological activity was done using the methods described in our previous study.6) PC12 cells (20000) were seeded in each well of a 24-well microplate and incubated under a humidified atmosphere of 5% CO2 at 37°C. After 24 h, the medium was replaced with 1 mL of serum-free Dulbecco’s modified Eagle’s medium (DMEM) containing a sample or DMSO (0.5%). NGF (40 ng/mL) was used as a positive control. The morphological changes of the cells were observed under a microscope. Approximately, 100 cells were watched from a randomly chosen area. A cell having neurite outgrowth longer than the diameter of cell body was determined as a positive cell. Significant differences between each groups were tested by ANOVA, followed by two-tailed multiple Student–Newman–Keuls t-tests using SPSS biostatistics software (IBM; Armonk, NY, U.S.A.). Values of p<0.05 were considered significant. Independent experiments were repeated for three times. Each value represents as the mean±standard error of the mean (S.E.M.) of three replicates. ** or *** indicates significant differences relative to the control at p<0.01 or p<0.001, respectively.
Western BlotWestern blot analysis was performed as previously described.10) Briefly, 1.5×106 cells were incubated in 6 cm dish with 15d (ABG-199) at a concentration of 0.1 µM for 0, 2, 4, 6, 8 or 10 h. The cells were lysed in 150 µL lysis buffer and then sonicated. The supernatant was removed after centrifugation at 12000 rpm for 15 min and protein concentration was determined by Bio-Rad protein assay kit (Bio-Rad Lab.; CA, U.S.A.). The cells were treated with 40 ng/mL NGF as positive control. A 15 µg of protein was loaded into sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and electrophoresised, then protein was transferred onto the polyvinylidene difluoride (PVDF) membrane. The membrane was blocked and incubated with anti-phospho-p44/42 mitogen-activated protein kinase (MAPK) (Thr202/Tyr204) rabbit polyclonal antibody (Cell Signaling Technology; MA, U.S.A.) for one hour at room temperature. The horseradish peroxidase-conjugated secondary antibody (Beijing ComWin Biotech Co., Ltd., Beijing, China) was incubated with membrane subsequently. The bands were developed with enhanced chemiluminescent reaction (Beijing ComWin Biotechnology) and analyzed by the software-Image J (National Institutes of Health, MD, U.S.A.).