Abstract
Two new flavonol glycosides were isolated from the leaves of Siparuna gigantotepala. Their structures were determined to be kaempferol 3-O-β-xylopyranosyl-(1→2)-α-arabinofuranoside (1) and kaempferol 3,7-di-O-methyl-4′-O-α-rhamnopyranosyl-(1→2)-β-glucopyranoside (2). In addition, three known flavonol glycosides, rutin (3), kaempferol 3-O-rutinoside (4), and kaempferol 3,7-di-O-methyl-4′-O-rutinoside (5), and three flavonol aglycones, quercetin (6), kaempferol 3,7-dimethyl ether (7), and kaempferol 3,7,4′-trimethyl ether (8), were also isolated and are reported here for the first time in this species. The structures of compounds 1 and 2 were established on the basis of their LC-MS and one- and two-dimensional (1D)- and (2D)-NMR spectroscopic analyses, combined with acid methanolysis and silylation of sugar moieties for GC-MS. Evaluation of the antioxidant activity, conducted in the 96-well plate format, showed that the flavonoids isolated possess strong 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical-scavenging activity and moderate oxygen radical absorption capacity.
Siparuna gigantotepala RENNER & HAUSNER (Siparunaceae, formerly Monimiaceae),1,2) a dioecious shrub or treelet first described in 1995 in humid primary pluvial forests in western Colombia and Ecuador at elevations of 20–1500 m, differs from all other Andean species in the elongate and oblong tepals persisting in fruit.3) Its strong aroma of lemon in the fruiting season in August gives its common name “Limón de monte.” Although there are no reports of secondary metabolites for this species, for some other of this genus have been reported oxoaporphine alkaloids in S. gilgiana4) and S. apiosyce,5) S. nicaraguensis, S. patelliformis, S. dresslerana,6) isoquinoline alkaloids and a cinnamic acid derivative in S. sessiliflora KUNTH A. DC,7) free and glycosylated flavonoids from Siparuna guianensis,8,9) Siparuna apiosyce,10) sesquiterpenoids from Siparuna pauciflora11) and essential oils for S. nicaraguensis, S. thecaphora,12) and S. guianensis.13)
In this study, five flavonoid glycosides including two new compounds (1 and 2) and rutin14,15) (3), kaempferol 3-O-rutinoside16) (4), kaempferol 3,7-di-O-methyl-4′-O-rutinoside (5)10) and the aglycons quercetin (6), kaempferol 3,7-dimethyl ether17) (7), and kaempferol 3,7,4′-trimethyl ether18) (8), were isolated from a methanolic extract of S. gigantotepala leaves. The structures of isolated flavonoids were elucidated through LC-electrospray ionization-mass spectrometry (ESI-MS), one- and two-dimensional (1D and 2D) homonuclear and heteronuclear NMR spectroscopical data analysis, combined with acid methanolysis and silylation of sugar moieties for gas chromatography mass spectrometry GC-MS. The antioxidant evaluation was performed by 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging activity and oxygen radical absorption capacity (ORAC) assays.
Results and Discussion
The methanol extract of S. gigantotepala showed potent antioxidant activity in one preliminary assay conducted to determine free radical scavenging activity of some “Reserva Nacional Forestal Bosque de Yotoco” plants, located in the southwest of Colombia.19) This result prompted us to search for antioxidant compounds of the methanol extract of S. gigantotepala leaves, a species not previously investigated. The methanol extract was sequentially liquid–liquid partitioned with ethyl acetate and n-butanol. Then butanolic fraction upon column chromatography on DIAION HP-20 and MCI gel with water–methanol stepwise gradient led to obtain an antioxidant active fraction Sg 2.2.3 (see Experimental).
On the basis of these results, a targeted isolation of these fraction constituents was performed by preparative chromatography obtaining the new flavonoid glycosides 1 and 2 (Chart 1).
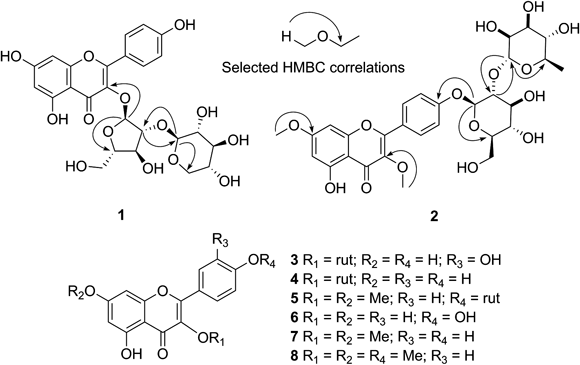
Chart 1. Flavonoids and Flavonoid Glycosides Isolated from Siparuna gigantotepala Leaves
The molecular formula of 1 was concluded to be C25H26O14 on the basis of ESI-MS corresponding to the molecular weight of 550 Da, which was evidenced by parent ions at m/z 551 [M+H]+ and 549 [M−H]− in the positive and negative ion modes respectively. High resolution electrospray ionization mass spectrometry (HR-ESI-MS) also confirmed the molecular mass as indicated in experimental section. In addition, the positive ESI-MS spectrum showed an ion at m/z 287 corresponding to a protonated kaempferol, obtained by loss of neutral fragments corresponding to a mass 264, suggesting the presence of two pentose residues. The strong IR absorption at 3383 cm−1 indicated the presence of multiple hydroxyl groups.
The 13C-NMR spectrum confirmed the presence of 25 carbons (Table 1). The 1H-NMR spectrum showed resonance for two meta-coupled aromatic protons at δ 6.21 (d, J=2.0 Hz, H-6) and 6.45 (d, J=2.0 Hz, H-8); and four ortho-coupled aromatic protons at δ 6.90 (d, J=9.0 Hz, H-3′, 5′) and 7.98 (d, J=9.0 Hz, H-2′, H-6′) consistent with a AA′BB′ spin system that indicated a p-substituted B ring. The flavonoid moiety of 1 was thus determined as kaempferol.
Table 1.
13C- and
1H-NMR Data (
J in Hz) of Isolated Compounds from
S. gigantotepala (400 MHz, δ ppm, in DMSO-
d6)
| C-n | 1 | 2 |
|---|
| 13C | 1H | 13C | 1H |
|---|
| 2 | 156.4 | — | 155.4 | — |
| 3 | 133.2 | — | 138.5 | — |
| 4 | 177.6 | — | 178.3 | — |
| 4a | 104.0 | — | 105.4 | — |
| 5 | 161.2 | — | 161.0 | — |
| 6 | 98.8 | 6.21, d (2.0) | 98.0 | 6.35, d (2.2) |
| 7 | 164.4 | — | 165.4 | |
| 8 | 93.8 | 6.45, d (2.0) | 92.6 | 6.76, d (2.2) |
| 8a | 156.9 | — | 156.5 | |
| 1′ | 120.6 | — | 123.5 | — |
| 2′ | 130.8 | 7.98, d (9.0) | 130.2 | 8.05, d (9.0) |
| 3′ | 115.5 | 6.90, d (9.0) | 116.1 | 7.16, d (9.0) |
| 4′ | 160.1 | — | 159.4 | |
| 5′ | 115.5 | 6.90, d (9.0) | 116.1 | 7.16, d (9.0) |
| 6′ | 130.8 | 7.98, d (9.0) | 130.2 | 8.05, d (9.0) |
| 3-OCH3 | — | — | 60.0 | 3.81, s |
| 7-OCH3 | — | — | 56.2 | 3.84, s |
| 1″ | 106.2 | 5.79, br s | 98.0 | 5.16, d (7.6) |
| 2″ | 88.8 | 4.32, dd (3.1, 2.3) | 76.6 | 3.54, dd (9.0, 7.6) |
| 3″ | 75.7 | 3.90, dd (6.3, 3.1) | 77.5 | 3.48, ft (9.0) |
| 4″ | 86.1 | 3.52, m | 70.7 | 3.20, ft (9.4) |
| 5″A | 60.5 | 3.26, dd (12.1, 5.4) | 77.1 | 3.52, m |
| 5″B | | 3.32, dd (12.1, 3.8) | — | — |
| 6″A | — | — | 60.7 | 3.51, dd (11.8, 8.5) |
| 6″B | — | — | | 3.72, m |
| 1‴ | 102.5 | 4.35, d (7.7) | 100.7 | 5.13, d (2.0) |
| 2‴ | 73.3 | 2.95, dd (9.0, 7.7) | 69.9 | 3.71, d (3.2, 2.0) |
| 3‴ | 76.7 | 3.09, ft (8.8) | 70.6 | 3.35, dd (9.3, 3.2) |
| 4‴ | 69.5 | 3.28, m | 72.0 | 3.23, ft (9.4) |
| 5‴ | 65.9 | 3.02, dd (11.5, 9.9) | 68.5 | 3.83, m |
| | 3.67, dd (11.5, 5.3) | — | — |
| 6‴ | — | — | 18.2 | 1.21, d (6.3) |
The 1H-NMR spectrum of 1 also exhibited a series of sugar signals at δ 2.95–4.32, along with two signals at δ 5.79 (br s, H-1″) and 4.35 (d, 7.7 Hz, H-1‴) corresponding to anomeric protons of both pentose residues. The heteronuclear multiple bond connectivity (HMBC) correlation between the anomeric proton H-1″ with the carbon at δ 133.2 (C-3) suggested the 3-O-glycosidation, and the interglycosidic linkage was determined to be 1→2 by the HMBC correlation from H-1‴ to the carbon at δ 88.8 (C-2″) and H-2″ at δ 4.32 with the carbon at δ 102.5 (C-1‴). Long range correlation from H-1″ to the signal at δ 86.1 (C-4″) and between H-5‴ at δ 3.67 with the anomeric carbon (C-1‴) at δ 102.5, suggest a furanose and a pyranose respectively; also supported by the signals at δ 60.5 and 65.9 corresponding to the carbons C-5″ and C-5‴ in 13C-NMR spectrum. The assignment of sugar signals from heteronuclear single quantum coherence (HSQC) and HMBC along with the coupling constants analysis from J-resolved indicated the first sugar moiety to be α-arabinofuranose. The anomeric proton H-1‴ (J=7.7 Hz) in a chair conformation for pyranose ring corresponded to axial–axial relationship with proton H-2‴ and the false triplet at δ 3.09 (J=8.8 Hz) indicates that the protons H-1‴–4‴ are axial, reducing the number of possible structures to β-xylopyranose. The sugar units were determined by GC-MS of methanolysate and silylated derivatives, confirming them as xylopyranose and arabinofuranose. Thus the structure of 1 was assigned as kaempferol 3-O-β-xylopyranosyl-(1→2)-α-arabinofuranoside.
The positive HR-ESI-MS spectrum of 2 showed an [M+H]+ ion at 623.1786, suggesting the molecular formula C29H34O15; which agree with [M+HCOO]− ion at 667.1716 in negative HR-ESI-MS spectrum of 2. The ESI-MS2 spectrum in positive mode showed an ion [(M+H)−146]+ at m/z 477, suggesting the neutral loss of a rhamnose moiety; while in negative mode spectrum the peak at m/z 313 due to the dimethyl kaempferol and the loss fragment corresponding to the diglycoside rhamnosyl-hexoside. Its IR spectrum revealed the carbonyl group of a 4-pyrone (1653 cm−1) and a hydroxyl group (3360 cm−1).
The 1H-NMR spectrum of 2 exhibited signals for a kaempferol moiety, two singlets at δ 3.81 and 3.84 ascribable to the methoxyl groups located on the carbons at δ 138.5 (C-3) and 165.4 (C-7), respectively as evidenced by the HMBC correlations. Moreover, two signals at δ 5.16 (d, J=7.6 Hz, H-1″) and 5.13 (d, J=2.0 Hz, H-1‴), which, on the basis of total correlation spectroscopy (TOCSY), correlation spectroscopy (COSY), J-resolved, HSQC and HMBC experiments, were assigned to the anomeric protons of β-glucopyranosyl and α-rhamnopyranosyl units respectively, which were confirmed by comparing the GC-MS data of the methanolysis/silylation products with those of authentic standards as shown in experimental section. The 13C chemical shifts for glucopyranosyl moiety (Table 1) are in accordance with those in the literature.20) The HMBC spectrum showed correlation between the anomeric proton H-1″ with the carbon C-4′ at δ 159.4 and correlation between the anomeric proton H-1‴ with the carbon C-2″ at δ 76.6, suggesting an 1→2 interglycosidic linkage. Thus, compound 2 is the new kaempferol 3,7-O-dimethyl-4′-O-α-rhamnopyranosyl-(1→2)-β-glucopyranoside.
In addition to compounds 1 and 2, rutin14,15) (3), kaempferol 3-O-rutinoside16) (4), kaempferol 3,7-di-O-methyl-4′-O-rutinoside (5)21) and the aglycons quercetin (6), kaempferol 3,7-dimethyl ether17) (7), and kaempferol 3,7,4′-trimethyl ether18) (8) were also isolated in this study. Their structures were identified by comparison of their spectroscopical data with those of published values. This work constitutes the first report of flavonols in Siparuna gigantotepala.
The antioxidant activity of the extracts and isolated compounds was tested by the DPPH assay and expressed as concentration of sample required to scavenge 50% DPPH free radicals (FRS50) value (Table 2). The results showed that DPPH values of extracts decrease with fractionation, showing an increase in the active compounds concentration. The flavonoid 3 was comparable to those of quercetin and vitamin C, flavonoids 4 and 1 displayed moderate activity, while flavonoids 5 and 2 were weakly active, suggesting that ortho hydroxyl groups and free hydroxyl groups at C-7 improves the radical scavenging activity.
Table 2. Antioxidant Activity
a) of Methanolic Fractions and Compounds from
Siparuna gigantotepala Leaves
| DPPH | ORAC (µM TE µM−1)c) |
|---|
| FRS50 (mg/L)b) | FRS50 (µM)b) |
|---|
| Sg 2 | 32±1.0 | | NTd) |
| Sg 2.2 | 28±1.1 | | NTd) |
| Sg 2.2.3 | 10.4±0.53 | | NTd) |
| Sg 2.2.5 | 47.1±0.98 | | NTd) |
| 1 | 16.1±0.62 | 29±1.1 | 0.44±0.064 |
| 2 | 53±2.2 | 86±3.5 | 0.19±0.010 |
| 3 | 5.2±0.28 | 8.5±0.5 | 1.17±0.10 |
| 4 | 15.4±0.29 | 25.9±0.5 | 0.5±0.11 |
| 5 | 70.7±0.80 | 114±1.3 | 0.07±0.011 |
| Quercetin (6) | 3.3±0.28 | 10.9±0.9 | 1.2±0.13 |
a) Data were expressed as the mean±S.D. b) Concentration of sample required to scavenge 50% DPPH free radicals. c) Micromoles of Trolox equivalents per micromole of sample. d) No tested.
Similar behavior was observed in the ORAC assay. The differences between the results of these two methods were possibly due to their different reaction mechanisms and measurements.22) The kaempferol glycosides were less active than quercetin glycosides in both the DPPH and ORAC assays, suggesting that antioxidant activity increases with the presence of hydroxyl groups and particularly with the presence of a B-ring catechol group.23)
Experimental
General Experimental ProceduresOptical rotations were measured with a JASCO model P-2000 series digital polarimeter. UV-visible spectra were recorded using a Shimadzu UV-1700 PharmaSpec spectrophotometer. Infrared spectra were measured in a FTIR affinity-1 spectrophotometer Shimadzu Corp. (Kyoto, Japan) equipped with Attenuated Total Reflectance (ATR) accessory. 1H- and 13C-NMR spectra were recorded in DMSO-d6 with a Bruker Avance III 400 spectrometer at 400 and 100 MHz, respectively. Fluorescence spectra and measurements were obtained in a JASCO FP-8500 Series spectrofluorometer, operated by JASCO Spectra Manager ver. 2.0. The HPLC analysis was performed in a Shimadzu Corp. LC-2010 instrument (Kyoto, Japan), equipped with a CBM-20 A system controller, a DGU-20 A degasser, a LC-20AD pump, a CTO-20AC column oven, a SIL-20AHT autosampler and an SPD-M20A PDA detector. Analytical HPLC was performed on a Restek Pinnacle DB C18 5 µm (250×4.6 mm) column. Semipreparative HPLC was performed on a Restek Pinnacle II C18 5 µm (250×21.2 mm). Positive and negative ESI-MS were recorded on a Shimadzu LCMS-2010 equipment and HR-MS were obtained on a Shimadzu LCMS-IT-time-of-flight (TOF) equipment. GC/MS analysis of the trimethylsilylated derivatives was performed in a Shimadzu model GCMS QP2010 Ultra system, operated in electron impact (EI) mode at 70 eV in full scan mode (range 35–700 m/z) with a scan speed of 1428 scans s−1. Separation of the methyl silylated monosacharides was performed on a Restek (Bellefont, PA, U.S.A.) chemically bonded Rtx-5MS fused-silica capillary column (30 m×0.25 mm i.d.×0.25 µm film thickness) with temperature program as follows: 150°C for 2 min, ramp to 250°C at 3°C min−1, and hold at 250°C for 3 min. The injector was operated at 280°C in split mode at 1 : 10 split ratio. Helium with a purity of a 99.999% was used as the carrier gas at a flow rate of 1.0 mL min−1, Flow Control by linear velocity at 38.0 cm/s. Samples (1.0 µL) were injected with an AOC-20i+s autosampler, 10 µL Hamilton micro syringe (Reno, NV, U.S.A.).
Plant MaterialLeaves of S. gigantotepala were collected inside the “Reserva Nacional Forestal Bosque de Yotoco,” Valle del Cauca, Colombia; which is heritage of the “Universidad Nacional de Colombia.” A sample was deposited at the Herbarium CUVC “Luis Sigifredo Espinal Tascón,” Universidad del Valle (CUVC 51736 Voucher), and it was identified by Dr. Philip A. Silverstone-Sopkin.
Extraction and IsolationThe air-dried leaves of S. gigantotepala (500 g) underwent ultrasonic assisted sequential extraction in chloroform (3 times×15 min) followed by methanol (3 times×15 min). Both solutions were dried under reduced pressure to obtain the corresponding extracts, chloroformic (Sg 1, 48.58 g, 9.7%) and methanolic (Sg 2, 35.34 g, 7.1%).
A portion of methanolic extract Sg 2 (25.34 g) was separated by sequential liquid–liquid partition to obtain ethyl acetate (Sg 2.1, 1.3 g, 5.13%), n-BuOH (Sg 2.2, 4.1 g, 16.2%) and aqueous (Sg 2.3, 18.0 g, 71%) extracts.
The butanolic extract Sg 2.2 (4.0 g) was separated in a stepwise gradient of water–MeOH (10–100% MeOH, increment of 20% in each step) on a DIAION HP-20 column (30 cm×5 cm i.d.) to get five fractions (recovery 81%). Flavonoid-rich fraction Sg 2.2.3 (500 mg) was chromatographed on MCI-Gel CHP20P (30 cm×2 cm i.d.) with a gradient of water–MeOH (30–100% MeOH, increments of 5% in each step) to obtained fifteen fractions (Recovery 90.8%). Fractions 6, 8, 9, 10 and 15, were further purified by preparative HPLC RP18 (H3PO4 (aq) 0.05%–isopropyl alcohol (IPA), 95 : 5, 92 : 8, 90 : 10, 88 : 12 and 80 : 20). Fraction 6 yielded 4 (8.2 mg); fraction 8 yielded 3 (23.8 mg); fraction 9 yielded 3 (12.5 mg), 4 (7.4 mg) and 1 (12.8 mg); fraction 10 yielded 4 (10.7 mg) and 6 (15.1 mg) and fraction 15 yielded 5 (13.8 mg) and 2 (21.3 mg).
Methanolysis and Silylation of Flavonoid GlycosidesFlavonoid glycosides and authentic monosaccharides (Sigma-Aldrich), including D-(−)-arabinose, D-(−)-lyxose, D-(−)-fructose, L-(−)-fucose, L-rhamnose, D-(+)-xylose, D-(−)-ribose, D-(+)-mannose, D-(+)-galactose and D-(+)-glucose (1 mg) were heated with 2 N methanolic HCl (0.5 mL) for 3 h in a boiling water bath to give 1-O-methyl glycosides. After removal of the solvent by evaporation in vacuo, 1 mL of acetonitrile was added and free hydroxyl groups were trimethylsilylated with 100 µL of N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) and trimethylchlorosilane (TMCS) (99 : 1) as reagent (Supelco) for 15 min at 70°C.
Kaempferol 3-O-β-Xylopyranosyl-(1→2)-α-arabinofuranoside (1)Yellow powder; [α]D25 +73.3 (c=0.80), MeOH; UV (MeOH) λmax (log ε) 270 (3.91), 300 (3.67), 356 (3.70); +NaOMe 273 (3.97), 315 (3.68), 388 (3.89); +NaOAc 271 (3.98), 300 (3.66), 357 (4.07); +NaOAc+H3BO3 264 (3.92), 344 (4.11); +AlCl3 271 (3.95), 297 (3.55), 336 (3.97), 397 (3.70); +AlCl3+HCl 271 (3.94), 296 (3.56), 337 (4.00), 398 (3.07) nm; IR (ATR) νmax cm−1: 3383 (OH), 1652 (carbonyl), 1608, 1503, 1466, 1160, 1030; 1H- and 13C-NMR, see Table 1; [+] ESI-MS m/z 551 [M+H]+, 287 [C15H11O6]+; [−] ESI-MS m/z 549 [M−H]−. [+] HR-ESI-MS m/z 573.1188 (Calcd for C25H26NaO14+, 573.1215), [−] HR-ESI-MS m/z 595.1278 (Calcd for [C25H26O14+HCOO]−, 595.1305). Methyl 2,3,4-tri-O-(trimethylsilyl)-α/β-D-xylopyranoside, tR 7.646/7.978, RI 1645/1666, MS m/z (%): 73 (100), 133 (33.12/30.31), 147 (32.81/25.29), 204 (58.25/66.00), 217 (43.93/47.27). Methyl 2,3,5-tri-O-(trimethylsilyl)-α/β-arabinofuranoside, tR 5.93/6.119, RI 1537/1550, MS m/z (%): 73 (100), 133 (21.25/23.64), 147 (14.62/20.24), 204 (18.87/22.21), 217 (38.59/40.93).
Kaempferol 3,7-Di-O-methyl-4′-O-α-rhamnopyranosyl-(1→2)-β-glucopyranoside (2)Yellow powder; [α]D25 −70.6 (c=1.3), MeOH; UV (MeOH) λmax (log ε) 268 (4.33), 338 (4.21); +NaOMe 268 (4.32), 338 (4.18); +NaOAc 268 (4.26), 338 (4.17); +NaOAc+H3BO3 268 (4.30), 338 (4.18); +AlCl3 280 (4.24), 300 (3.88), 342 (4.28), 400 (3.72); +AlCl3+HCl 280 (4.24), 300 (3.88), 340 (4.17), 400 (3.53) nm; IR (ATR) νmax cm−1: 3360 (OH), 1653 (carbonyl), 1597, 1498, 1348, 1165, 1041; 1H- and 13C-NMR, see Table 1; positive ESI-MS m/z 623.15 [M+H]+, 477.10 [(M+H)−146]+; negative ESI-MS 621.10 [M−H]−, 312.95 [(M−H)−146−162]−; [+] HR-ESI-MS m/z 623.1945 (Calcd for C29H35O15+ [M+H]+ 623.1970), [−] HR-ESI-MS m/z 667.1854 (Calcd for [C29H34O15+HCOO]−, 667.1880). Methyl 2,3,4-tri-O-(trimethylsilyl)-α/β-L-rhamnopyranoside, tR 6.39/6.599, RI 1568/1581, MS m/z (%): 73 (100), 133 (18.31/24.31), 147 (13.85/23.71), 204 (49.36/51.91), 217 (8.27/15.04). Methyl 2,3,4,6-tetrakis-O-(trimethylsilyl)-α/β-D-glucopyranoside, tR 11.764/12.103, RI 1873/1891, MS m/z (%): 73 (100), 133 (22.26/29.91), 147 (18.15/13.67), 204 (45.97/47.18), 217 (12.85/12.74).
Measurement of DPPH Radical Scavenging ActivityThe DPPH free radical scavenging of pure compounds were measured in a 96-well microplates format.24) Briefly, 100 µL of diluted samples at different concentrations (1–1024 mg/L), was mixed with 100 µL of 132 mg/L DPPH solution in methanol, the plate was incubated at room temperature, after 1 h, measurements of absorbance were done at 520 nm in a microplate reader (Metertech, AccuReader M 965+). Ascorbic acid and quercetin were used as positive controls and ran in parallel. The results were expressed as a percentage of radical scavenging activity (%FRS) according to the equation:
where
Ac is the absrobance of DPPH radicals without sample or positive controls and
As is the absrobance of DPPH radicals with sample or positive control. The efficient concentration of samples and positive controls that inhibits FRS
50 was calculed and expressed as mg/L.
Measurement of Oxygen Radical Absorbance Capacity (ORAC)ORAC assay was carried out according to method described by Huang et al.25) with some modifications. The assay was performed in a 96-well plate. Sodium fluorescein was dissolved in phosphate buffer solution (PBS) (75 mM, pH 7.4) to obtain a stock solution of 4 µM. The working solution (0.08 µM) was prepared by subsequent dilution in PBS. A 10 mL solution of 2,2-azobis(2-amidinopropane)dihydrochloride (AAPH) was prepared at a concentration of 250 µM (67.8 mg/mL in PBS). For each session of measurements, a standard curve of Trolox was plotted (0.625–5 µM). Trolox solutions were prepared in PBS. A blank (PBS) was run with each assay. Samples were diluted in PBS at different concentrations (0.625–5 µM) from stock solutions at 1 mg/mL in dimethyl sulfoxide (DMSO). Sample (50 µL) was mixed with sodium fluorescein (100 µL) and incubated for 10 min at 37°C. The reaction was started by the addition of AAPH (50 µL) and fluorescence was measured immediately during 120 min in an espectrofluorometer JASCO FP-8500 (λexc 490 nm, λem 514 nm). The area under the curve (AUC) of each sample was calculated relative to the initial value (the fluorescence intensity at 0 min), and the blank value was subtracted. The regression coefficient between AUC and antioxidant concentration was calculated for all samples (r2>0.93). The results expressed as µmol of Trolox Equivalent (TE) per µmol of sample. Further positive control measurements were performed with quercetin.
Statistical AnalysisAll the reported values represent the mean±standard deviation (S.D.) of experiments performed in triplicate and statistically processed in GraphPad Prism 6.0.
Acknowledgments
Authors are grateful to “Universidad del Valle” and COLCIENCIAS for the financial support through the Grant CI7852, CT-557-2011. H.G. Torres C., thanks to “Universidad Nacional de Colombia” (Palmira) for the study commission to perform his doctoral studies.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Renner S. S., Hausner G., Novon, 10, 134–143 (2000).
- 2) Renner S. S., Hausner G., Novon, 15, 202–206 (2005).
- 3) Renner S. S., Hausner G., Novon, 5, 61–70 (1995).
- 4) Chiu S. Y. C., Dobberstein R. H., Fong H. H. S., Farnsworth N. R., J. Nat. Prod., 45, 229–230 (1982).
- 5) Fischer D. C. H., Gonçalves M. I., Oliveira F., Alvarenga M. A., Fitoterapia, 70, 322–323 (1999).
- 6) Gerard R. V., MacLean D. B., Antonio T. M., Phytochemistry, 25, 2155–2156 (1986).
- 7) González G. F. P., Archila E. G., Rev. Cubana Plant. Med., 17, 65–72 (2012).
- 8) Negri G., de Santi D., Tabach R., Rev. Bras. Farmacogn., 22, 1024–1034 (2012).
- 9) Leitão G. G., El-Adji S. S., Araújo Lopes de Melo W., Leitão S. G., Brown L., J. Liq. Chromatogr. R. T., 28, 2041–2051 (2005).
- 10) Leitão G. G., Soares S. S. V., Brito T. D., Delle Monache F., de Barros T., Phytochemistry, 55, 679–682 (2000).
- 11) Jenett-Siems K., Kraft C., Siems K., Jakupovic J., Solis P. N., Gupta M. P., Bienzle U., Phytochemistry, 63, 377–381 (2003).
- 12) Vila R., Iglesias J., Canigueral S., Santana A. I., Solis P. N., Gupta M. P., J. Essential Oil Res., 14, 66–67 (2002).
- 13) Viana F. A., Andrade-Neto M., Pouliquen Y. B. M., Uchoa D. E. A., Sobral M. M. S. Z., De Morais S. M., J. Essential Oil Res., 14, 60–62 (2002).
- 14) Kazuma K., Noda N., Suzuki M., Phytochemistry, 62, 229–237 (2003).
- 15) Adjé F. A., Lozano Y. F., Le Gernevé C., Lozano P. R., Meudec E., Adima A. A., Gaydou E. M., Ind. Crops Prod., 37, 303–310 (2012).
- 16) Leong C. N. A., Tako M., Hanashiro I., Tamaki H., Food Chem., 109, 415–420 (2008).
- 17) Herz W., Fitzhenry B., Anderson G. D., Phytochemistry, 12, 1181–1182 (1973).
- 18) Stevens J. F., Hart H. T., Wollenweber E., Phytochemistry, 39, 805–813 (1995).
- 19) Torres Castañeda H., Colmenares de Vélez A. J., Isaza Martínez J. H., Revista de Ciencias, 17, 35–44 (2014).
- 20) da Silva B. P., Bernardo R. R., Parente J. P., Phytochemistry, 53, 87–92 (2000).
- 21) Leitão G. G., Soares S. S. V., Brito M., Monache F. D., de Barros T., Phytochemistry, 55, 679–682 (2000).
- 22) Villaño D., Fernández-Pachón M. S., Troncoso A. M., García-Parrilla M. C., Anal. Chim. Acta, 538, 391–398 (2005).
- 23) Van Hoyweghen L., Karalic I., Van Calenbergh S., Deforce D., Heyerick A., J. Nat. Prod., 73, 1573–1577 (2010).
- 24) Sdiri S., Bermejo A., Aleza P., Navarro P., Salvador A., Food Res. Int., 49, 462–468 (2012).
- 25) Huang D., Ou B., Hampsch-Woodill M., Flanagan J. A., Prior R. L., J. Agric. Food Chem., 50, 4437–4444 (2002).
