Regular Articles
Synthesis and in Vitro Antifungal Activities of Novel Benzamide Derivatives Containing a Triazole Moiety
2016 Volume 64 Issue 6 Pages 616-624
Details
2016 Volume 64 Issue 6 Pages 616-624
The study reported the synthesis and antifungal activities in vitro against six phytopathogenic fungi of 17 novel N-[2-hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]benzamide derivatives. All the target compounds were synthesized and elucidated by means of MS, high resolution (HR)-MS, IR, 1H- and 13C-NMR analysis. The results showed that almost all the derivatives exhibited good activities against each of the tested fungi at the concentration of 50 µg/mL. Among them, 6h displayed excellent activity against Alternaria alternata with the median effective concentration value (EC50) of 1.77 µg/mL, superior to myclobutanil (EC50=6.23 µg/mL), a commercial fungicide with broad-spectrum bioactivities for plant protection and high-efficiency. Compound 6k showed the broadest antifungal spectrum, demonstrating positive activities against the corresponding fungi with EC50 values ranging from 0.98 to 6.71 µg/mL. Furthermore, 6e to 6i revealed good activities against Alternaria solani with EC50 values of 1.90, 4.51, 7.07, 2.00 and 5.44 µg/mL, respectively. The preliminary analysis of structure–activity relationship (SAR) demonstrated that the presence of F or Cl on the benzene ring remarkably improved the activity, while the introduction of 4-OMe or CF3 group decreased the activity in varying degrees. Thus, the present results strongly suggest that N-[2-hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]benzamide derivatives should be promising candidates for the development of novel antifungal agents in the effective control of phytopathogenic fungi.
Diseases caused by phytopathogenic fungi have been one of the most severe concerns in a wide range of field.1) These fungi can invade all the tissues of a plant at its any growth stage, thereby leading to significant losses in the yield and quality of agricultural products.1–3) Moreover, many fungal plant pathogens could produce mycotoxins, the fungal secondary metabolic products that may pose a threat to animal and human health.1,3,4) Therefore, the effective control of fungal diseases is extremely essential for agricultural production, food safety and human health worldwide.2,3)
To date, chemical control is still considered as an efficient measure for preventing and controlling these diseases,2) and various synthesized fungicides have been extensively developed and commercially available.5–7) The representatives are triazole fungicides, such as triadimefon, triadimenol and so on (Fig. 1). As an important class of sterol biosynthesis inhibitors (SBI), they are of great significance to protect crops, and have achieved a unique position in the chemical control of fungal diseases since their discovery in the late 1960 s.5,7) However, intensive application of these fungicides has caused the appearance of resistant strains, which results in diminishing their effectiveness.8)
Another important type is amide fungicide, like carboxin and boscalid, etc. (Fig. 1), whose common target is mitochondrial respiratory chain enzyme complex II (succinate dehydrogenase, SDH). Because of their features of high efficiency and broad spectrum, these fungicides attracted great attention of many researchers, and have been used for crop protection currently.6,7)

Considering the positive effects of the two types of fungicides described above, in this study, we combined the active structure of triazole fungicides with benzamide or naphthamide groups, both of which have different modes of action,6) to design and synthesize a series of novel benzamide derivatives containing a 1,2,4-triazole moiety (Fig. 2), aiming for obtaining compounds possessing more potent antifungal activities, especially against resistant fungi, as well as establishing their structure–activity relationships (SAR). Herein, 3,3-dimethyl-1-(1H-1,2,4-triazol-1-yl)-2-butanone (also called 1,2,4-triazol-1-ylpinacolin for short), an important intermediate to synthesize diverse triazole fungicides, was chosen as a starting material based on relevant SARs which have been established through enormous researches.6)

Commercially available 1,2,4-triazol-1-ylpinacolin (1) was used as a starting material to react with trimethylsulfoxonium iodide (TMSOI), known as sulfur ylide,9) in the presence of tetrabutylammonium iodide (TBAI) (phase transfer catalyst), and 50% potassium hydroxide (KOH) aqueous solution (reaction catalyst) in toluene to prepare compound 2 in 78% yield. Next, compound 2 then reacted with sodium azide in N,N-dimethylformamide (DMF) in the presence of NH4Cl to yield compound 3, followed by the treatment with triphenylphosphine in dichloromethane to afford compound 4 in 91% yield. Afterwards, 1-amino-3,3-dimethyl-2-(1H-1,2,4-triazol-1-yl)methyl-2-butanol (compound 5) as a key intermediate was obtained in 91% yield through hydrolyzing compound 4 using sodium hydroxide aqueous solution (2 mol/L) in methanol. Finally, compound 5 reacted with a variety of benzoyl chlorides or naphthoyl chlorides in dried dichloromethane in the presence of triethylamine, used as the deacid reagent to accelerate acylation reaction, to obtain the target compounds 6a to q (Chart 1).

R=H; 2-Me; 3-Me; 4-Me; 2-F; 3-F; 4-F; 2-Cl; 3-Cl; 4-Cl; 2,4-diCl; 2-CF3; 3-CF3; 4-CF3; 4-OMea. TMSOI, KOH, TBAI, toluene, 70°C, 78% yield; b. NaN3, NH4Cl, DMF, 60–70°C; c. PPh3, CH2Cl2, r.t., 91% yield; d. NaOH, CH3OH, reflux, 91% yield; e. ArCOCl, Et3N, dry CH2Cl2, 0°C to r.t., 56–97% yield.
The in vitro antifungal activities of synthetic compounds 6a to q against six plant pathogenic fungi (Alternaria solani, Magnaporthe grisea, Alternaria alternata, Curvularia lunata, Fusarium graminearum, Fusarium solani) at the concentration of 50 µg/mL were screened by the mycelium linear growth rate method.1,3,10–12) Compound 5, 1-amino-3,3-dimethyl-2-(1H-1,2,4-triazol-1-yl)methyl-2-butanol, a key intermediate, was regarded as the reference compound, whose antifungal activity was also assayed by the same methodology, and myclobutanil, a commercial fungicide, was used as the positive control. The results are displayed in Table 1.
| Compound | Average inhibition rate±S.D. (%) (n=3) | ||||||
|---|---|---|---|---|---|---|---|
| No. | R | M. grisea | A. solani | F. graminearum | F. solani | C. lunata | A. alternata |
| 6a | H | 45±5.8 jka) | 77±1.6 e | 53±2.7 b | 44±5.1 gh | 57±8.7 cde | 73±10.2 c |
| 6b | 2-Me | 41±13.8 jk | 86±3.3 bcde | 53±4.3 b | 53±11.0 efgh | 38±6.4 fg | 57±6.3 de |
| 6c | 3-Me | 51±3.0 ij | 65±5.1 f | 27±3.4 c | 58±6.1 cdefg | 46±8.1 ef | 47±7.8 ef |
| 6d | 4-Me | 59±10.4 ghi | 89±5.0 abc | 0±11.6 f | 62±6.1 bcdef | 50±2.8 def | 58±3.7 de |
| 6e | 2-F | 97±1.1 a | 87±5.1 abcd | 94±1.6 a | 65±4.9 bcde | 78±3.3 b | 96±1.1 ab |
| 6f | 3-F | 72±4.0 def | 92±10.2 ab | 24±5.6 cd | 72±6.7 bc | 59±5.8 cd | 63±4.7 cd |
| 6g | 4-F | 82±5.9 bcd | 97±3.1 a | 55±7.2 b | 71±5.1 bc | 77±3.6 b | 86±6.0 b |
| 6h | 2-Cl | 51±8.2 ij | 91±3.3 abc | 58±4.0 b | 49±9.7 fgh | 46±3.9 ef | 87±6.5 b |
| 6i | 3-Cl | 77±2.1 cde | 91±4.8 ab | 18±11.6 cde | 67±7.8 bcd | 64±6.8 c | 64±3.1 cd |
| 6j | 4-Cl | 35±10.7 kl | 80±3.1 cde | 22±10.8 cde | 45±9.9 gh | 44±9.2 f | 63±8.6 cd |
| 6k | 2,4-diCl | 97±2.9 a | 93±1.1 ab | 98±0.0 a | 76±0.9 b | 94±9.8 a | 87±8.3 b |
| 6l | 2-CF3 | 57±3.4 hi | 45±3.2 g | 14±4.3 de | 40±11.6 h | 6±10.4 h | 27±5.6 g |
| 6m | 3-CF3 | 64±7.9 fgh | 92±3.6 ab | 2±9.5 f | 55±7.8 defgh | 40±8.0 fg | 58±6.9 de |
| 6n | 4-CF3 | 29±8.8 l | 51±1.9 g | 10±6.4 ef | 43±8.8 h | 30±3.9 g | 44±14.6 f |
| 6o | 4-OMe | 70±3.2 defg | 43±2.9 g | 47±8.5 b | 53±7.9 efgh | 38±6.5 fg | 40±4.6 f |
| 6p | 69±2.7 efgh | 83±2.1 cd | 56±2.0 b | 71±3.9 bc | 44±4.6 f | 48±6.5 ef | |
| 6q | 83±0.0 bc | 78±3.7 de | 50±7.6 b | 44±9.7 gh | 46±6.4 ef | 97±1.7 ab | |
| 5 | 27±7.6 l | 61±16.3 f | −20±10.4 g | 18±7.8 i | −1±10.0 h | 24±6.7 g | |
| Myclobutanil | 85±4.1 bc | 100±0.0 a | 100±0.0 a | 100±0.0 a | 100±0.0 a | 100±0.0 a | |
a) The differences between data with the different lowercase letters within a column are significant for the tested fungus (p<0.05), which were carried out by Duncan’s multiple comparison.
It was clearly seen from Table 1 that almost all the derivatives (6a to q) exhibited better inhibitory activities against each of the test fungi than the reference compound 5, except for 6n and o against A. solani. Especially, compound 6e showed excellent activity against M. grisea with the inhibition rate of 97%, which was significantly higher than that of myclobutanil (85%) (p<0.05), a commercial fungicide. Compared with other target compounds, 6k showed broader antifungal spectrum, which showed inhibitory activities against M. grisea, A. solani, F. graminearum, C. lunata and A. alternata with inhibition rates of 97, 93, 98, 94 and 87%, respectively. Generally, most of these compounds were more active against A. solani, with inhibition rates exceeding 85%, followed by A. alternata, M. grisea, F. graminearum, C. lunata and F. solani.
Antifungal ToxicityIn order to further explore the antifungal potency, the compounds with inhibition rates over 85% based on the screening results displayed above (Table 1) were further examined to determine their median effective concentrations (EC50) against corresponding strains of fungi. The assay method was the same as that mentioned above.1,3,11,13) Compound 5 and myclobutanil were used as the reference compound and positive control, respectively. Toxicity regression equations for concentration–effect of the compounds and their corresponding EC50 values are listed in Tables 2 to 6.
| Compound | Regression equationa) | R2 | EC50 | 95% CIb) | Linear scope (µg/mL) |
|---|---|---|---|---|---|
| µg/mL | |||||
| 6b | y=1.7553x+3.7095 | 0.9986 | 5.44 | 5.32–5.56 | 0.78–50 |
| 6d | y=2.014x+2.6402 | 0.9104 | 14.85 | 3.47–63.63 | 0.78–50 |
| 6e | y=1.3223x+4.6314 | 0.9826 | 1.90 | 1.33–2.71 | 0.78–50 |
| 6f | y=0.9828x+4.3571 | 0.9434 | 4.51 | 1.56–13.06 | 0.78–50 |
| 6g | y=1.2143x+3.9683 | 0.9030 | 7.07 | 1.72–29.10 | 0.78–50 |
| 6h | y=1.0392x+4.6862 | 0.9644 | 2.00 | 1.36–2.96 | 1.56–25 |
| 6i | y=1.1698x+4.1399 | 0.9798 | 5.44 | 4.26–6.93 | 1.56–50 |
| 6k | y=1.3181x+5.0132 | 0.9678 | 0.98 | 0.62–1.53 | 0.78–25 |
| 6m | y=1.576x+3.9673 | 0.9379 | 4.52 | 1.25–16.37 | 0.78–50 |
| 5 | y=1.3727x+1.2428 | 0.9043 | 545.87 | 191.71–1554.33 | 25–1600 |
a) y: Probability of average inhibition rate; x: lg[concentration (µg/mL)]. b) Confidence interval of EC50 (µg/mL) at 95% probability.
Table 2 indicated that all of the 9 tested compounds exhibited good activities against A. solani. Their EC50 values were 0.98–14.85 µg/mL, which were much smaller than that of reference compound 5 (EC50=545.87 µg/mL), and the values of 8 out of 9 tested compounds were below 10.0 µg/mL. As to A. alternata, all 5 tested compounds possessed excellent activities, and their EC50 values ranged from 1.77 to 28.88 µg/mL (Table 3). Especially, 6h showed the most potent activity, with an EC50 value of 1.77 µg/mL, superior to myclobutanil, the positive control (EC50=6.23 µg/mL), and the reference compound 5 (EC50=1215.52 µg/mL). From Tables 4 and 5, it was clearly seen that both 6e and k displayed great activities against M. grisea with EC50 values of 5.22, 2.35 µg/mL, and F. graminearum with EC50 values of 4.64, 4.48 µg/mL, respectively, and their activities were much higher than that of the reference compound 5 (EC50= >1600, 416.18 µg/mL, respectively). Besides, 6k revealed definite activity against C. lunata, with an EC50 value of 2.62 µg/mL (Table 6), which was much stronger than the reference compound 5 with EC50 value >1600 µg/mL.
| Compound | Regression equationa) | R2 | EC50 | 95% CIb) | Linear scope (µg/mL) |
|---|---|---|---|---|---|
| µg/mL | |||||
| 6e | y=1.2997x+3.6954 | 0.9381 | 10.09 | 6.37–15.97 | 3.13–50 |
| 6g | y=0.6395x+4.1686 | 0.9513 | 19.96 | 9.52–41.84 | 0.78–50 |
| 6h | y=0.6722x+4.8326 | 0.9314 | 1.77 | 0.46–6.79 | 0.78–50 |
| 6k | y=0.8298x+4.3138 | 0.9642 | 6.71 | 3.84–11.74 | 0.78–50 |
| 6q | y=0.7658x+3.8815 | 0.9770 | 28.88 | 19.22–43.38 | 0.78–50 |
| 5 | y=1.1444x+1.4698 | 0.9062 | 1215.52 | 272.72–5418.54 | 25–1600 |
| Myclobutanil | y=1.1909x+4.0542 | 0.9300 | 6.23 | 4.34–8.93 | 1.56–12.5 |
a) y: Probability of average inhibition rate; x: lg[concentration (µg/mL)]. b) Confidence interval of EC50 (µg/mL) at 95% probability.
| Compound | Regression equationa) | R2 | EC50 | 95% CIb) | Linear scope (µg/mL) |
|---|---|---|---|---|---|
| µg/mL | |||||
| 6e | y=1.6951x+3.7835 | 0.9347 | 5.22 | 1.99–13.72 | 0.78–50 |
| 6k | y=0.6682x+4.7527 | 0.9802 | 2.35 | 1.75–3.14 | 0.78–50 |
| Myclobutanil | y=1.3975x+4.9163 | 0.9767 | 1.15 | 0.81–1.62 | 0.78–25 |
a) y: Probability of average inhibition rate; x: lg[concentration (µg/mL)]. b) Confidence interval of EC50 (µg/mL) at 95% probability.
| Compound | Regression equationa) | R2 | EC50 | 95% CIb) | Linear scope (µg/mL) |
|---|---|---|---|---|---|
| µg/mL | |||||
| 6e | y=0.7988x+4.4678 | 0.9942 | 4.64 | 4.40–4.89 | 1.56–50 |
| 6k | y=1.1403x+4.2578 | 0.9650 | 4.48 | 2.53–7.93 | 0.78–50 |
| 5 | y=1.0002x+2.3802 | 0.9731 | 416.18 | 281.72–614.86 | 25–1600 |
a) y: Probability of average inhibition rate; x: lg[concentration (µg/mL)]. b) Confidence interval of EC50 (µg/mL) at 95% probability.
| Compound | Regression equationa) | R2 | EC50 | 95% CIb) | Linear scope (µg/mL) |
|---|---|---|---|---|---|
| µg/mL | |||||
| 6k | y=1.25x+4.4762 | 0.9411 | 2.62 | 1.19–5.79 | 1.56–50 |
a) y: Probability of average inhibition rate; x: lg[concentration (µg/mL)]. b) Confidence interval of EC50 (µg/mL) at 95% probability.
The results described above indicated that all the tested compounds were obviously more active than the reference compound 5. It was worth mentioning that compound 6k had a broad antifungal spectrum, exhibiting excellent activities against almost all the test fungi. In addition, the strain of A. solani was more susceptible to these tested compounds than other fungi strains. These results were basically consistent with the screening results in Table 1.
SARBy comparing the results of EC50 values in Tables 2 to 6, with the inhibition rates of all the target compounds at 50 µg/mL in Table 1, it was obviously seen that almost all of the benzamide or naphthamide derivatives (6a to q) possessed higher activity against each of the test fungi than their key intermediate 5, a compound containing triazole only, which implied that it was effective to increase the antifungal activities of compounds by combining the active structure of triazoles with benzamide group.
Furthermore, the results described above revealed that the type and position of the substituents on benzene ring had great influences upon the antifungal activity. Compared with the unsubstituted compound 6a (R=H), the presence of 2-F, 4-F or 2,4-diCl was beneficial for the improvement of the activities against all the tested fungi. Moreover, the effects of other substituents on the activity depended on their positions on benzene ring and the species of fungi (Fig. 3).
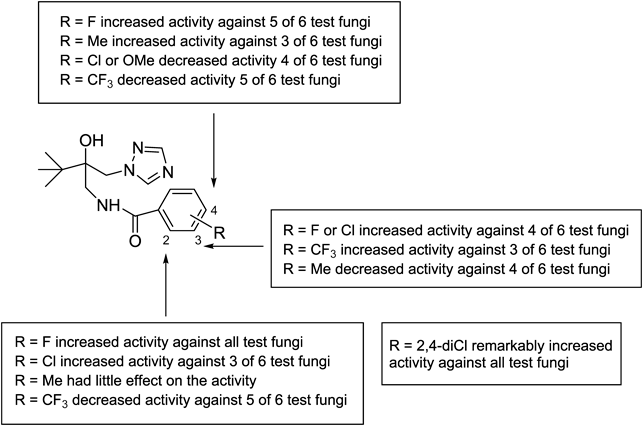
For M. grisea, the presence of a methyl group at the 2, 3 or 4 position on benzene ring had little effect on the activity, while the introduction of a fluorine atom remarkably improved the activity, especially 2-fluorinated compound 6e (EC50=5.22 µg/mL). For the four chlorinated compounds 6h to k, 6k (R=2,4-diCl) possessed the highest activity (EC50=2.35 µg/mL), followed by 6i (3-Cl), h (2-Cl), and j (4-Cl). A similar trend was observed in trifluoromethyl-substituted compounds, the order of the activity was 3->2->4-substituted isomer. However, the introduction of 4-methoxyl group slightly increased the activity compared with 6a (R=H). As to A. solani, although most of the target compounds exhibited good inhibitory activity in varying degrees, the presence of 3-Me, 4-OMe, 2-CF3 or 4-CF3 decreased the activity. Regarding F. solani, the introduction of methyl, fluoro, chloro, trifluoromethyl or 4-methoxyl slightly increased the activity compared with the unsubstituted 6a. However, the introduction of methyl, chloro, trifluoromethyl or 4-methoxyl decreased the activity against F. graminearum, whereas, only 2-fluorinated (6e) as well as 2,4-dichlorinated (6k) isomers revealed definite activity. The similar trends were observed on C. lunata and A. alternata. In general, for all the test fungi, the presence of methyl (6b to d), trifluoromethyl (6l to n) or 4-methoxyl (6o) on benzene ring was of less importance to improve the activity, while the introduction of fluorine or chlorine atom was beneficial for improving the activity in varying degrees, especially 2-F and 2,4-diCl derivatives exhibited excellent activity.
In addition, naphthamide derivatives (6p to q) showed lower activities than benzamides (6b to o) against almost all the test fungi, with the exception of 6q against A. alternata with an EC50 value of 28.88 µg/mL.
In conclusion, the present study reported the synthesis of a series of novel N-[2-hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]benzamide or naphthamide derivatives, and subsequent evaluation of their antifungal activities in vitro against six plant pathogenic fungi. Furthermore, the preliminary SAR was discussed. Most of the target compounds were found to have activities in varying degrees at 50 µg/mL for 72 h post-treatment, and showed much higher activities against all the test fungi than their intermediate compound 5. Among them, compound 6h displayed excellent activity against A. alternata with an EC50 value of 1.77 µg/mL, superior to myclobutanil (EC50=6.23 µg/mL), a commercial fungicide. Compound 6k had the broadest antifungal spectrum, showing good activities against most of the tested fungi, whereas 6e to i exhibited definite activity against A. solani. SAR analysis demonstrated that the type and position of substituents on the benzene ring had significant effects on the activity. Generally, the presence of 2-F, 3-F, 4-F, 2-Cl, 3-Cl or 2,4-diCl was of importance for increasing the activity, while the introduction of 4-OMe or CF3 group decreased the activity. Thus, the present results strongly suggest that the target compounds, i.e., benzamide derivatives containing 1,2,4-triazole moiety, should be potential candidates for the development of novel fungicides for the effective control of phytopathogenic fungi.
Myclobutanil, a commercial fungicide, was purchased from Yi Fang Biotechnology Co., Ltd. (Zhejiang, China). 3,3-Dimethyl-1-(1H-1,2,4-triazol-1-yl)-2-butanone, the starting material, was purchased from Jiangsu Yancheng Chemical Factory (Jiangsu, China). Other reagents and solvents were obtained locally with analytical grade and used without further purification.
ApparatusMelting points (mp) were determined on an XT-4 micro-melting point apparatus and uncorrected. Infrared (IR) spectra were performed on a Bruker TENSOR 27 spectrometer with KBr disks. 1H- and 13C-nuclear magnetic resonance (NMR) were recorded on a Bruker Avance III 500 MHz instrument with tetramethylsilane (TMS) as an internal standard. Chemical shift (δ values) and coupling constants (J values) were given in ppm and Hz, respectively. Electrospray ionization (ESI)-MS were measured on a Thermo Fisher LCQ Fleet instrument. High resolution (HR)-MS were carried out with Thermo Scientific LTQ Orbitrap instrument.
Synthesis of Compound 2According to the reported methods9,14,15) with modification, the aqueous solution of potassium hydroxide (50%, 50 mL) was slowly added in the mixture of 3,3-dimethyl-1-(1H-1,2,4-triazol-1-yl)-2-butanone (1) (36.7 g, 0.22 mol), trimethylsulphoxonium iodide (TMSOI; 55 g, 0.25 mol), TBAI (0.5 g) and toluene (120 mL) with stirring, and then the reaction mixture was heated to 70°C for several hours until the reaction was completed according to TLC detection. The mixture was extracted with 120 mL ethyl acetate, and the aqueous phase was extracted again with ethyl acetate (3 × 60 mL). The combined organic phase was washed with saturated saline solution (3×50 mL), and finally dried over anhydrous Na2SO4. After filtration, the solution was evaporated in vacuum to yield 32.5 g yellow oil, followed by adding diethyl ether (25 mL), and then recrystallized to yield compound 2.
2-t-Butyl-2-(1H-1,2,4-triazol-1-yl)methyl Oxirane (2)Colorless crystal in 78% yield, mp 49.9–50.7°C. 1H-NMR (500 MHz, CDCl3) δ: 8.08 (1H, s), 7.90 (1H, s), 4.66 (1H, d, J=15.1 Hz), 4.48 (1H, d, J=15.1 Hz), 2.71 (1H, d, J=3.6 Hz), 1.88 (1H, d, J=3.6 Hz), 1.04 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 151.5, 144.8, 62.2, 48.9, 47.5, 32.8, 25.9.
Synthesis of Compound 3Compound 2 (18.0 g, 0.1 mol), sodium azide (11.7 g, 0.18 mol) and ammonium chloride (6.4 g, 0.12 mol) were fully dissolved in 100 mL DMF. Afterwards, the mixture was stirred and heated slowly to 60–70°C for 1 h. The solution was then poured into water (200 mL), and extracted with chloroform (3×80 mL). The combined organic layer was washed with water (3×150 mL), and finally dried over anhydrous Na2SO4. After filtration, the solvent was removed in vacuum to yield 1-azido-3,3-dimethyl-2-(1H-1,2,4-triazol-1-yl)methyl-2-butanol (3) as pale yellow oil. The crude intermediate 3 was directly used in the following step.
Synthesis of Compound 4A solution of compound 3 (22.4 g, 0.1 mol) and triphenylphosphine (26.2 g, 0.1 mol) in dichloromethane (100 mL) was stirred at room temperature until the reaction was completed according to TLC detection. After removal of the solvent, the residue was recrystallized in ethyl acetate to get compound with satisfactory yield.
1-(Triphenylphosphoranylidene)amino-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]-2-butanol (4)White crystal in 91% yield, mp 150.7–151.8°C. 1H-NMR (500 MHz, CDCl3) δ: 8.30 (1H, s), 7.62 (1H, s), 7.43–7.51 (15H, m), 6.14 (1H, s), 4.36 (1H, d, J=13.9 Hz), 4.30 (1H, d, J=13.9 Hz), 3.07 (1H, t, J=11.3 Hz), 2.96–2.99 (1H, m), 0.90 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 150.1, 144.6, 132.3 (d, J=9.3 Hz), 131.5 (d, J=2.5 Hz), 130.5 (d, J=98.0 Hz), 128.5 (d, J=11.7 Hz), 73.3, 55.2, 45.3, 37.2, 25.3.
Synthesis of Compound 5A mixture of compound 4 (27.4 g, 0.06 mol) and sodium hydroxide aqueous solution (2 mol/L, 80 mL) in methanol (200 mL) was stirred under reflux for 3 h, and then cooled to room temperature before removal of the solvent. The residue was added to toluene (100 mL) and water (100 mL), followed by acidification with hydrochloric acid (10%) to pH 2–3. After the mixture was partitioned, aqueous phase was collected, washed with toluene (3×80 mL), and alkalized to pH 11–12 with anhydrous sodium carbonate. Subsequently, the aqueous phase was extracted with dichloromethane (3×80 mL), and dried over anhydrous Na2SO4. After filtration and removal of the solvent, the residue was dissolved with ethyl acetate and recrystallized in a mixed solution of ethyl acetate and n-hexane to yield compound.
1-Amino-3,3-dimethyl-2-(1H-1,2,4-triazol-1-yl)methyl-2-butanol (5)White lamellar crystal in 91% yield. IR (KBr) cm−1: 3387 (O-H), 3335, 1511 (N-H), 1598 (C=N). 1H-NMR (500 MHz, CDCl3) δ: 8.23 (1H, s), 7.92 (1H, s), 5.13 (1H, s), 4.26–4.32 (2H, m), 2.94 (1H, d, J=13.4 Hz), 2.73 (1H, d, J=13.4 Hz), 1.01 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 151.5, 145.4, 72.7, 55.2, 41.3, 37.1, 25.2. Positive ESI-MS m/z: 199.2 [M+1]+, 197.0 [M−1]+. HR-MS [M+H]+: Calcd for C9H19N4O+ 199.1553. Found 199.1552.
Synthesis of Compounds 6a to qAccording to the reported method16) with modification, compound 5 (0.4 g, 2 mmol) and triethylamine (0.3 mL) were dissolved in the dried dichloromethane (5 mL) under stirring in ice bath. Then a solution of benzoyl/naphthoyl chloride (2 mmol) in dried dichloromethane (5 mL) was dropwise added into the above mixture. The resulting solution was stirred for 0.5 h at 0°C, and warmed to room temperature with stirring until the reaction was completed according to TLC detection. The reaction mixture was washed with water (3×10 mL), and the collected organic phase was dried with anhydrous Na2SO4. After filtration and removal of the solvent, the residue was purified by column chromatography on silica gel using petroleum ether–ethyl acetate (1 : 3, v/v) as eluent.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]benzamide (6a)Colorless oil in 93% yield. IR (KBr) cm−1: 1643 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.94 (1H, s), 7.61 (1H, s), 7.59 (1H, d, J=1.4 Hz), 7.49 (1H, tt, J=7.6 Hz, 1.2 Hz), 7.40 (2H, t, J=7.9 Hz), 6.47 (1H, s), 4.42 (1H, d, J=14.3 Hz), 4.31 (1H, d, J=14.3 Hz), 4.24 (1H, s), 3.90–3.94 (1H, m), 3.55–3.59 (1H, m), 1.07 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 169.7, 151.7, 145.5, 133.2, 131.9, 128.6, 126.9, 77.2, 53.5, 43.6, 37.8, 25.3. Positive ESI-MS m/z: 325.4 [M+Na]+. HR-MS [M+H]+: Calcd for C16H23N4O2+ 303.1816. Found 303.1815.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-2-methylbenzamide (6b)Pale yellow lamellar solid in 56% yield, mp 117.5–118°C. IR (KBr) cm−1: 1631 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.22 (1H, s), 7.82 (1H, s), 7.28–7.31 (1H, m), 7.16–7.19 (2H, m), 7.11 (1H, d, J=7.8 Hz), 6.11 (1H, s), 4.31–4.41 (2H, m), 4.25 (1H, s), 3.95–3.99 (1H, m), 3.49–3.53 (1H, m), 2.37(3H, s), 1.07 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 172.7, 151.7, 145.4, 136.5, 135.0, 131.2, 130.3, 126.6, 125.8, 77.3, 53.4, 43.5, 37.8, 25.3, 19.9. Positive ESI-MS m/z: 339.3 [M+Na]+. HR-MS [M+H]+: Calcd for C17H25N4O2+ 317.1972. Found 317.1970.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-3-methylbenzamide (6c)Colorless oil in 56% yield. IR (KBr) cm−1: 1643 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.94 (1H, s), 7.43 (1H, s), 7.36 (1H, d, J=6.3 Hz), 7.29 (2H, d, J=6.3 Hz), 6.42 (1H, s), 4.41 (1H, d, J=14.4 Hz), 4.31 (1H, d, J=14.4 Hz), 4.25 (1H, s), 3.91–3.95 (1H, m), 3.53–3.57 (1H, m), 2.38 (3H, s), 1.07 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 170.0, 151.7, 145.5, 138.5, 133.1, 132.7, 128.5, 127.7, 123.8, 77.2, 53.5, 43.6, 37.8, 25.3, 21.3. Positive ESI-MS m/z: 339.3 [M+Na]+. HR-MS [M+H]+: Calcd for C17H25N4O2+ 317.1972. Found 317.1969.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-4-methylbenzamide (6d)Colorless oil in 61% yield. IR (KBr) cm−1: 1642 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.19 (1H, s), 7.93 (1H, s), 7.49 (2H, d, J=8.2 Hz), 7.20 (2H, d, J=8.0 Hz), 6.39 (1H, s), 4.41 (1H, d, J=14.4 Hz), 4.29–4.32 (2H, m), 3.91–3.95 (1H, m), 3.52–3.57 (1H, m), 2.38 (3H, s), 1.07 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 169.7, 151.7, 145.5, 142.5, 130.3, 129.3, 126.9, 77.2, 53.5, 43.6, 37.8, 25.3, 21.5. Positive ESI-MS m/z: 339.4 [M+Na]+. HR-MS [M+H]+: Calcd for C17H25N4O2+ 317.1972. Found 317.1969.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-2-fluorobenzamide (6e)White granular crystal in 63% yield, mp 136.2–137.8°C. IR (KBr) cm−1: 1655 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.16 (1H, s), 7.97 (1H, td, J=7.9 Hz, 1.8 Hz), 7.91 (1H, s), 7.44–7.48 (1H, m), 7.20–7.24 (1H, m), 7.06–7.11 (1H, m), 6.87 (1H, s), 4.41 (1H, d, J=14.3 Hz), 4.31 (2H, d, J=14.3 Hz), 3.96–3.99 (1H, m), 3.57–3.62 (1H, m), 1.08 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 165.8, 161.6, 159.6 (d, J=248.9 Hz), 151.8, 145.2, 133.8 (d, J=9.3 Hz), 131.9 (d, J=1.8 Hz), 124.8 (d, J=3.3 Hz), 119.9 (d, J=11.3 Hz), 116.2, 116.0 (d, J=24.6 Hz), 53.5, 43.7, 37.8, 25.3. Positive ESI-MS m/z: 343.4 [M+Na]+. HR-MS [M+H]+: Calcd for C16H22FN4O2+ 321.1721. Found 321.1720.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-3-fluorobenzamide (6f)Colorless oil in 93% yield. IR (KBr) cm−1: 1647 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.95 (1H, s), 7.32–7.40 (3H, m), 7.17–7.21 (1H, m), 6.51 (1H, s), 4.42 (1H, d, J=14.4 Hz), 4.32 (1H, d, J=14.4 Hz), 4.09 (1H, s), 3.86–3.90 (1H, m), 3.56–3.61 (1H, m), 1.08 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 168.2, 163.7, 161.7 (d, J=247.9 Hz), 151.8, 145.5, 135.6 (d, J=7.3 Hz), 130.3 (d, J=7.3 Hz), 122.3 (d, J=3.6 Hz), 118.8 (d, J=21.3 Hz), 114.3 (d, J=23.2 Hz), 77.2, 53.4, 43.6, 37.8, 25.2. Positive ESI-MS m/z: 343.4 [M+Na]+. HR-MS [M+H]+: Calcd for C16H22FN4O2+ 321.1721. Found 321.1720.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-4-fluorobenzamide (6g)Colorless oil in 84% yield. IR (KBr) cm−1: 1644 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.93 (1H, s), 7.60–7.62 (2H, m), 7.08 (2H, t, J=8.6 Hz), 6.40 (1H, s), 4.42 (1H, d, J=14.4 Hz), 4.31 (1H, d, J=14.4 Hz), 4.17 (1H, s), 3.88–3.92 (1H, m), 3.54–3.58 (1H, m), 1.08 (9H, s); 13C-NMR (125 MHz, CDCl3) δ: 168.6, 166.0, 164.0 (d, J=252.6 Hz), 151.7, 145.5, 129.4 (d, J=3.6 Hz), 129.2 (d, J=8.2 Hz), 115.6 (d, J=21.8 Hz), 77.2, 53.5, 43.6, 37.8, 25.2. 13C-NMR (125 MHz, DMSO-d6) δ: 167.3, 165.12, 163.12 (d, J=250.8 Hz), 149.4, 144.5, 129.3, 128.6 (d, J=8.9 Hz), 114.2 (d, J=22.3 Hz), 75.6, 52.3, 42.3, 36.6, 23.6. Positive ESI-MS m/z: 343.3 [M+Na]+. HR-MS [M+H]+: Calcd for C16H22FN4O2+ 321.1721. Found 321.1720.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-2-chlorobenzamide (6h)Colorless oil in 65% yield. IR (KBr) cm−1: 1647 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.21 (1H, s), 7.85 (1H, s), 7.47 (1H, dd, J=7.5, 1.5 Hz), 7.34–7.39 (2H, m), 7.28–7.32 (1H, m), 6.52 (1H, s), 4.34–4.43 (2H, m), 4.14 (1H, s), 3.91–3.95 (1H, m), 3.59–3.63 (1H, m), 1.06 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 168.9, 151.8, 145.3, 134.0, 131.7, 130.7, 130.3, 130.0, 127.1, 77.3, 53.2, 43.7, 37.8, 25.3. Positive ESI-MS m/z: 359.4 [M+Na]+. HR-MS [M+H]+: Calcd for C16H22ClN4O2+ 337.1426. Found 337.1424.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-3-chlorobenzamide (6i)Colorless crystal in 95% yield, mp 122.2–123.4°C. IR (KBr) cm−1: 1645 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.23 (1H, s), 7.99 (1H, s), 7.65 (1H, t, J=1.7 Hz), 7.47–7.51 (2H, m), 7.38 (1H, t, J=7.9 Hz), 6.53 (1H, s), 4.45 (1H, d, J=14.4 Hz), 4.36 (1H, d, J=14.4 Hz), 4.10 (1H, s), 3.90–3.95 (1H, m), 3.59–3.63 (1H, m), 1.11 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 168.2, 151.8, 145.6, 135.0, 134.9, 131.9, 129.9, 127.4, 124.9, 77.1, 53.4, 43.6, 37.8, 25.2. Positive ESI-MS m/z: 359.3 [M+Na]+. HR-MS [M+H]+: Calcd for C16H22ClN4O2+ 337.1426. Found 337.1424.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-4-chlorobenzamide (6j)Colorless oil in 92% yield. IR (KBr) cm−1: 1642 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.93 (1H, s), 7.53 (2H, d, J=8.6 Hz), 7.38 (2H, d, J=8.6 Hz), 6.44 (1H, s), 4.42 (1H, d, J=14.4 Hz), 4.31 (1H, d, J=14.4 Hz), 4.13 (1H, s), 3.87–3.91 (1H, m), 3.55–3.59 (1H, m), 1.08 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 168.5, 151.7, 145.5, 138.2, 131.6, 128.9, 128.3, 77.2, 53.5, 43.6, 37.8, 25.2. Positive ESI-MS m/z: 359.4 [M+Na]+. HR-MS [M+H]+: Calcd for C16H22ClN4O2+ 337.1426. Found 337.1423.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-2,4-dichlorobenzamide (6k)Colorless oil in 95% yield. IR (KBr) cm−1: 1649 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.85 (1H, s), 7.44 (1H, d, J=8.3 Hz), 7.40 (1H, d, J=1.7 Hz), 7.28–7.30 (1H, m), 6.53 (1H, s), 4.41 (1H, d, J=14.4 Hz), 4.34 (1H, d, J=14.4 Hz), 4.05 (1H, s), 3.88–3.92 (1H, m), 3.58–3.63 (1H, m), 1.06 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.7, 151.8, 145.4, 137.2, 132.3, 131.6, 131.2, 130.2, 127.5, 77.3, 53.2, 43.7, 37.8, 25.3. Positive ESI-MS m/z: 393.4 [M+Na]+. HR-MS [M+H]+: Calcd for C16H21Cl2N4O2+ 371.1036. Found 371.1034.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-2-(trifluoromethyl)benzamide (6l)Colorless oil in 66% yield. IR (KBr) cm−1: 1651 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.23 (1H, s), 7.81 (1H, s), 7.68 (1H, d, J=7.6 Hz), 7.52–7.59 (2H, m), 7.28 (1H, s), 6.09 (1H, s), 4.37 (2H, q, J=14.4 Hz), 3.93–3.97 (2H, m), 3.52–3.56 (1H, m), 1.06 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 170.3, 151.7, 145.5, 134.7, 132.1, 130.2, 128.4, 127.4, 127.2 (d, J=31.8 Hz), 126.5 (d, J=5.1 Hz), 124.5, 122.4 (d, J=273.4 Hz), 77.3, 53.2, 43.8, 37.8, 25.3. Positive ESI-MS m/z: 393.4 [M+Na]+. HR-MS [M+H]+: Calcd for C17H22F3N4O2+ 371.1689. Found 371.1686.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-3-(trifluoromethyl)benzamide (6m)White crystal in 71% yield, mp 123.3–124.6°C. IR (KBr) cm−1: 1649 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.94 (1H, s), 7.89 (1H, s), 7.74–7.78 (2H, m), 7.55 (1H, t, J=7.8 Hz), 6.58 (1H, s), 4.43 (1H, d, J=14.4 Hz), 4.34 (1H, d, J=14.4 Hz), 4.01 (1H, s), 3.90–3.94 (1H, m), 3.58–3.62 (1H, m), 1.09 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.9, 151.8, 145.6, 134.1, 131.3, 131.1 (d, J=32.7 Hz), 130.1, 129.3, 128.5, 128.4 (d, J=3.6 Hz), 124.0 (d, J=3.6 Hz), 124.7, 122.6 (d, J=273.1 Hz), 77.1, 53.5, 43.7, 37.8, 25.2. Positive ESI-MS m/z: 393.4 [M+Na]+. HR-MS [M+H]+: Calcd for C17H22F3N4O2+ 371.1689. Found 371.1686.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-4-(trifluoromethyl)benzamide (6n)White granular crystal in 63% yield, mp 114.2–115.9°C. IR (KBr) cm−1: 1656 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.94 (1H, s), 7.69 (4H, q, J=8.4 Hz), 6.53 (1H, s), 4.43 (1H, d, J=14.4 Hz), 4.33 (1H, d, J=14.4 Hz), 4.01 (1H, s), 3.88–3.92 (1H, m), 3.58–3.63 (1H, m), 1.09 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 168.2, 151.8, 145.6, 136.6, 133.7, 133.4 (d, J=32.7 Hz), 127.4, 125.7 (q-like, J=3.7 Hz), 124.7, 122.5 (d, J=272.7 Hz), 77.2, 53.5, 43.6, 37.8, 25.2. Positive ESI-MS m/z: 393.4 [M+Na]+. HR-MS [M+H]+: Calcd for C17H22F3N4O2+ 371.1689. Found 371.1687.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-4-methoxybenzamide (6o)White crystal in 95% yield, mp 128.0–129.6°C. IR (KBr) cm−1: 1637 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.20 (1H, s), 7.94 (1H, s), 7.56 (2H, d, J=8.7 Hz), 6.89 (2H, d, J=8.7 Hz), 6.32 (1H, s), 4.41 (1H, d, J=14.4 Hz), 4.35 (1H, s), 4.30 (1H, d, J=14.3 Hz), 3.91–3.95 (1H, m), 3.83 (3H, s), 3.51–3.55 (1H, m), 1.07 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 169.3, 162.6, 151.7, 145.5, 128.8, 125.4, 113.8, 77.2, 55.4, 53.6, 43.58, 37.8, 25.3. Positive ESI-MS m/z: 355.4 [M+Na]+. HR-MS [M+H]+: Calcd for C17H25N4O3+ 333.1921. Found 333.1919.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-1-naphthamide (6p)Colorless oil in 97% yield. IR (KBr) cm−1: 1651 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.24 (1H, s), 8.20 (1H, d, J=8.6 Hz), 7.90 (1H, d, J=8.2 Hz), 7.85 (1H, d, J=7.2 Hz), 7.80 (1H, s), 7.50–7.55 (2H, m), 7.43 (1H, t, J=7.6 Hz), 7.34 (1H, d, J=7.3 Hz), 6.35 (1H, s), 4.42 (1H, d, J=14.4 Hz), 4.35 (1H, d, J=14.4 Hz), 4.28 (1H, s), 4.05–4.09 (1H, m), 3.58–3.62 (1H, m), 1.10 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 172.2, 151.8, 145.4, 133.7, 133.1, 131.1, 130.0, 128.4, 127.3, 126.5, 125.3, 125.0, 124.6, 77.4, 53.4, 43.7, 37.9, 25.3. Positive ESI-MS m/z: 375.4 [M+Na]+. HR-MS [M+H]+: Calcd for C20H25N4O2+ 353.1972. Found 353.1969.
N-[2-Hydroxy-3,3-dimethyl-2-[(1H-1,2,4-triazol-1-yl)methyl]butyl]-2-naphthamide (6q)White crystal in 86% yield, mp 142.5–143.3°C. IR (KBr) cm−1: 1649 (C=O). 1H-NMR (500 MHz, CDCl3) δ: 8.22 (1H, s), 8.11 (1H, s), 7.96 (1H, s), 7.91 (1H, d, J=7.8 Hz), 7.84–7.86 (2H, m), 7.63 (1H, dd, J=8.6, 1.7 Hz), 7.51–7.57 (2H, m), 6.58 (1H, s), 4.44 (1H, d, J=14.4 Hz), 4.33 (1H, d, J=14.4 Hz), 4.27 (1H, s), 3.97–4.02 (1H, m), 3.59–3.63 (1H, m), 1.09 (9H, s). 13C-NMR (125 MHz, CDCl3) δ: 169.8, 151.8, 145.6, 134.9, 132.6, 130.4, 129.1, 128.5, 127.9, 127.7, 126.8, 123.2, 77.3, 53.6, 43.7, 37.8, 25.3. Positive ESI-MS m/z: 375.4 [M+Na]+. HR-MS [M+H]+: Calcd for C20H25N4O2+ 353.1972. Found 353.1970.
Pharmacology in Vitro Antifungal Activity AssayThe antifungal activities in vitro of all the target compounds against six phytopathogenic fungi (A. solani, M. grisea, A. alternate, C. lunata, F. graminearum, F. solani) were assayed using the growth rate method1,3,10,11,13) with minor modification. The test fungi, provided by the Center of Pesticide Research, Northwest A&F University, China, and maintained on potato dextrose agar (PDA) medium slants were subcultured for 48 h in Petri dishes prior to testing and used for inoculation of fungal strains on PDA plates. All the target compounds were completely dissolved in acetone that served as the solvent to help these compounds to be diffused into the PDA medium. Sample solution (1 mg tested compound in 1 mL acetone) was completely mixed with the autoclaved PDA medium to afford the medium containing 50 µg/mL of target compound, and poured into Petri dishes in a laminar flow chamber. PDA medium containing the same amount of acetone was served as the blank control, and 50 µg/mL of myclobutanil, a commercial fungicide, was used as the positive control, respectively. When the medium in the plate was partially solidified, a 5 mm thick and 4 mm in diameter disc of fungus cut from beforehand subcultured Petri dishes was placed at the centre of semi-solid medium. The dishes were kept in an incubator at 28°C for 72 h. Each experiment was carried out in triplicate. The diameters (in mm) of inhibition zones were measured in three different directions and the growth inhibition rates were calculated according to the following formula and expressed as the mean±standard deviation (S.D.):
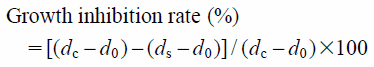 |
Based on the in vitro antifungal activity screening results, the more active compounds 6b, d, e, f, g, h, i, k, m and q were subject to determining their median effective concentration (EC50) according to the same method described above.1,3,10,11,13) A stock solution was prepared by dissolving the tested compounds in acetone, and then diluted by acetone using serial two-fold dilution method to obtain a series of stock solutions. Each stock solution was respectively mixed with the autoclaved PDA medium to prepare a series of media containing 50, 25, 12.5, 6.25, 3.125, 1.5625, 0.78125 µg/mL of the tested compounds. Meanwhile, the same volume of acetone was used as blank control. Each experiment was performed in triplicate. The average inhibition rate for each test was calculated and then transformed to the corresponding probit value. The concentration (µg/mL) of the compound was transformed to the corresponding logarithm value (lg C). Lg C values of each compound and its corresponding probit values were used to establish toxicity regression equation by the linear least-square fitting method.1,17) EC50 values and their confidence intervals at 95% probability were calculated from the corresponding toxicity regression equations.
Statistic AnalysisSPSS 22 statistical software was used to analyze the data and establish toxicity regression equations. Duncan multiple comparison test was performed on the data to evaluate significant difference between the activities of various compounds at the same concentration.
This work was supported by the Natural Science Research Program of Shaanxi Province of China (No. 2015JM3120) and the Research Fund for the Doctoral Program by Northwest A&F University (No. 2014BSJJ082).
The authors declare no conflict of interest.