Experimental
General Comments1H-NMR (500 MHz), 13C-NMR (125 MHz) spectra were determined on JEOL ECA-500 instrument. Chemical shifts for 1H-NMR were reported in parts per million downfields from tetramethylsilane (δ) as the internal standard and coupling constants were in hertz (Hz). The following abbreviations are used for spin multiplicity: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, br=broad. Chemical shifts for 13C-NMR were reported in ppm relative to the centerline of a triplet at 77.0 ppm for deuteriochloroform. HR-MS were obtained on a BRUKER DALTONICS micrOTOF (electrospray ionization (ESI)). IR spectra were recorded on a SHIMADZU IRPrestige-21. Optical rotations were measured on a JASCO P-1030 Polarimeter at RT using the sodium D line. Analytical TLC was performed on Merck precoated analytical plates, 0.25 mm thick, silica gel 60 F254. Preparative TLC separations were made on 7×20 cm plates prepared with a 0.25 mm layer of Merck silica gel 60 F254. Compounds were eluted from the adsorbent with 10% MeOH in chloroform. Column chromatography separations were performed on KANTO CHEMICAL Silica Gel 60 (spherical) 40–50 µm, Silica Gel 60 (spherical) 63–210 µm or Silica Gel 60 N (spherical, neutral) 63–210 µm. Reagents and solvents were commercial grades and were used as supplied with the following exceptions. 1) Dichloromethane, tetrahydrofuran and toluene: dried over molecular sieves 4A. 2) MeOH and acetonitrile: dried over molecular sieves 3A. All reactions sensitive to oxygen and/or moisture were conducted under an argon atmosphere.
2-Chloro-6-methoxypyridine (9)To a stirred solution of 2,6-dichloropyridine (8) (68.9 g, 466 mmol) in MeOH (500 mL) was added NaOMe (100 g, 1.86 mol) at room temperature. The resulting mixture was stirred at 60°C for 24 h. After cooling, the mixture was quenched with 2 M aqueous HCl, and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to afford 9 (66.9 g, quant) as a colorless oil.
9: IR (film, cm−1): 1599, 1585, 1560, 1468, 1410, 1302, 1265, 1152, 1024, 876, 789; 1H-NMR (CDCl3, 500 MHz) δ: 7.51 (t, J=7.37 Hz, 1H), 6.90 (d, J=7.37 Hz, 1H), 6.65 (d, J=7.37 Hz, 1H), 3.94 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 163.9, 148.4, 140.5, 116.2, 109.1, 54.0; HR-MS (ESI-time-of-flight (TOF)): Calcd for C6H7ClNO [(M+H)+] 144.0211. Found 144.0211.
Methyl 5-Formyl-6-methoxypicolinate (11)To a stirred solution of 9 (20.8 g, 145 mmol) in tetrahydrofuran (THF) (400 mL) was added t-BuLi (ca. 1.6 M solution in n-heptane, 100 mL, 160 mmol) at −78°C. After 1 h, DMF (33.8 mL, 435 mmol) was added at –78°C. After 30 min, the resulting mixture was stirred at room temperature for 30 min. Then the reaction mixture was quenched with 2 M aqueous HCl, and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude material including 10 was applied to the following reaction without further purification.
To a stirred suspension of the crude material including 10 and NaOAc (17.8 g, 218 mmol) in the 2 : 1 mixture of MeOH and toluene (total 450 mL) were added Pd(OAc)2 (651 mg, 2.90 mmol) and DPPF (2.41 g, 4.35 mmol) at room temperature. The resulting mixture was stirred under ordinary CO pressure (balloon) at 50°C for 23 h. Then the reaction mixture was quenched with 1 M aqueous HCl, and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=2 : 1) to afford 11 (27.5 g, 97%, 2 steps) as a colorless solid.
11: mp: 87–88°C; IR (film, cm−1): 1726, 1694, 1591, 1456, 1381, 1254, 1134; 1H-NMR (CDCl3, 500 MHz) δ: 10.4 (s, 1H), 8.22 (d, J=7.94 Hz, 1H), 7.79 (d, J=7.94 Hz, 1H), 4.17 (s, 3H), 4.00 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 188.6, 164.7, 164.0, 149.8, 138.6, 121.2, 118.5, 54.3, 53.0; HR-MS (ESI-TOF): Calcd for C9H9NO4Na [(M+Na)+] 218.0424. Found 218.0428.
3-Bromo-2-chloro-6-methoxypyridine (31)To a stirred solution of 9 (7.00 g, 48.8 mmol) in CH3CN (25 mL) was added NBS (13.0 g, 73.1 mmol) at room temperature. The resulting mixture was refluxed for 24 h. Then the reaction was quenched with saturated aqueous Na2S2O3 and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=98 : 2) to afford methoxypyridine 31 (6.83 g, 63%) as a colorless solid.
31: mp 64–65°C; IR (film, cm−1): 1584, 1551, 1466, 1408, 1344, 1306, 1256, 1155, 1121, 1022, 1009; 1H-NMR (CDCl3, 500 MHz) δ: 7.72 (d, J=8.50 Hz, 1H), 6.58 (d, J=8.50 Hz, 1H), 3.92 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 162.4, 147.3, 143.8, 110.9, 110.1, 54.3; HR-MS (ESI-TOF): Calcd for C6H6BrClNO [(M+H)+] 221.9316. Found 221.9314.
Butyl 3-Formyl-6-methoxypicolinate (13)To a stirred solution of 31 (6.83 g, 30.7 mmol) in THF (120 mL) was added i-PrMgCl·LiCl (ca. 1.0 M solution in THF, 32.2 mL, 32.2 mmol) at −20°C. After 2 h, DMF (7.2 mL, 92.1 mmol) was added dropwise at −20°C. The resulting mixture was stirred at room temperature for 30 min. The reaction mixture was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude material including 12 was applied to the following reaction without further purification.
To a suspension of the crude material including 12 and NaOAc (3.78 g, 46.1 mmol) in the 1 : 1 mixture of n-BuOH and toluene (total 120 mL) was added Pd(dppf)Cl2 (1.12 g, 1.54 mmol)at room temperature. The resulting mixture was stirred under ordinary CO pressure (balloon) at 100°C for 18 h. The reaction was quenched with saturated aqueous NH4Cl, filtered through a pad of Celite and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=2 : 1) to afford 13 (4.75 g, 65% 2 steps) as a yellow oil.
13: IR (film, cm−1): 2963, 2876, 1721, 1692, 1595, 1481, 1337, 1277, 1261, 1219, 1138, 1072, 1022; 1H-NMR (CDCl3, 500 MHz) δ: 10.39 (s, 1H), 8.18 (d, J=8.50 Hz, 1H), 6.94 (d, J=8.50 Hz, 1H), 4.44 (t, J=6.80 Hz, 2H), 4.06 (s, 3H), 1.83–1.76 (m, 2H), 1.54–1.45 (m, 2H), 0.99 (t, J=7.37 Hz, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 189.1, 166.1, 165.2, 150.7, 138.6, 125.9, 114.1, 66.3, 54.5, 30.5, 19.2, 13.7; HR-MS (ESI-TOF): Calcd for C12H15NO4Na [(M+Na)+] 260.0893. Found 260.0891.
Methyl 3-(Dimethoxymethyl)-6-methoxypicolinate (32)To a stirred solution of 13 (4.75 g, 20.0 mmol) in MeOH (100 mL) were added CH(OMe)3 (11 mL, 100 mmol) and 10-camphorsulfonic acid (CSA) (465 mg, 2.00 mmol) at room temperature. The resulting mixture was refluxed for 24 h. Then the reaction mixture was concentrated under reduced pressure. The residue was diluted with saturated aqueous NaHCO3 and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=9 : 1) to afford methoxypicolinate 32 (3.40 g, 70%) as a yellow oil.
32: IR (attenuated total reflectance (ATR), cm−1) 2951, 2832, 1730, 1597, 1479, 1321, 1250, 1217, 1109, 1070, 1051, 1026, 974, 831; 1H-NMR (CDCl3, 500 MHz) δ: 7.90 (d, J=8.50 Hz, 1H), 6.86 (d, J=8.50 Hz, 1H), 5.85 (s, 1H), 3.97 (s, 3H), 3.96 (s, 3H), 3.34 (s, 6H); 13C-NMR (CDCl3, 125 MHz) δ: 166.9, 163.3, 145.5, 138.3, 127.1, 113.0, 100.0, 53.8, 53.6, 52.6; HR-MS (ESI-TOF): Calcd for C11H15NO5Na [(M+Na)+] 264.0842. Found 264.0842.
tert-Butyl 3-Formyl-6-methoxypicolinate (14)To a stirred solution of 32 (500 mg, 2.07 mmol) in THF (4 mL) was added 1 M aqueous KOH (4.15 mL, 4.15 mmol). The resulting mixture was stirred at 40°C for 3 h. Then the mixture was concentrated under reduced pressure. The crude material including carboxylic acid (33) was applied to the following reaction without further purification.
To a stirred suspension of the crude material including 33 in CH2Cl2 (5 mL) were added NH4Cl (277 mg, 5.18 mmol) and N,N′-diisopropyl-O-tert-butylisourea (1.63 mL, 7.25 mmol) at room temperature. The resulting mixture was stirred at room temperature for 15 h. Then the reaction mixture was filtered and concentrated under reduced pressure. The crude material including tert-butyl ester 34 was applied to the following reaction without further purification.
To a stirred solution of the crude material including 34 in THF (2 mL) was added 1 M aqueous HCl (2 mL) at room temperature. The resulting mixture was stirred at the same temperature for 1.5 h. Then the reaction mixture was diluted with water and extracted with EtOAc. The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=4 : 1) to afford 14 (391 mg, 80%, 3 steps) as a colorless oil.
14: IR (film, cm−1): 2982, 1736, 1595, 1481, 1335, 1279, 1223, 1167, 1138, 1072, 1020, 845; 1H-NMR (CDCl3, 500 MHz) δ: 10.35 (s, 1H), 8.14 (d, J=8.50 Hz, 1H), 6.91 (d, J=8.50 Hz, 1H), 4.06 (s, 3H), 1.65 (s, 9H); 13C-NMR (CDCl3, 125 MHz) δ: 189.1, 166.1, 164.2, 152.3, 138.5, 125.1, 113.6, 83.9, 54.4, 28.1; HR-MS (ESI-TOF): Calcd for C12H15NO4Na [(M+Na)+] 260.0893. Found 260.0881.
(E)-Methyl 6-Methoxy-5-(2-nitrovinyl)picolinate (15)To a stirred solution of 11 (616 mg, 3.16 mmol) in CH3NO2 (10 mL) was added Et3N (319 mg, 3.16 mmol) at room temperature. The resulting mixture was stirring at the same temperature for 1.5 h. Then the reaction mixture was concentrated under reduced pressure. The crude material including nitroalcohol 35 was applied to the following reaction without further purification.
To a stirred solution of the crude material including 35 in CH2Cl2 (10 mL) were added Et3N (319 mg, 3.16 mmol) and MsCl (996 mg, 4.74 mmol) at 0°C. The resulting mixture was stirred at the same temperature for 3 h. Then the reaction mixture was quenched with saturated aqueous NaHCO3, and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, CHCl3–EtOAc=9 : 1) to afford 15 (750 mg, quant) as a pale yellow solid.
15: IR (film, cm−1): 1726, 1632, 1516, 1342, 1271; 1H-NMR (CDCl3, 500 MHz) δ: 8.02 (d, J=13.8 Hz, 1H), 7.98 (d, J=13.8 Hz, 1H), 7.87 (d, J=7.45 Hz, 1H), 7.79 (d, J=7.45 Hz, 1H), 4.19 (s, 3H), 3.99 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 164.8, 161.9, 147.3, 141.9, 140.9, 132.7, 118.8, 117.6, 54.6, 53.0; HR-MS (ESI-TOF): Calcd for C10H10N2O5Na [(M+Na)+] 261.0482. Found 261.0494.
(E)-tert-Butyl 6-Methoxy-3-(2-nitrovinyl)picolinate (16)To a stirred solution of 14 (1.48 g, 6.22 mmol) in CH3NO2 (30 mL) was added Et3N (1.72 mL, 12.4 mmol) at room temperature. The reaction mixture was stirred at the same temperature for 20 h. Then the reaction mixture was concentrated under reduced pressure. The crude material including nitroalcohol 36 was applied to the following reaction without further purification.
To a stirred solution of the crude material including 36 in CH2Cl2 (30 mL) were added Et3N (1.29 mL, 9.33 mmol) and MsCl (963 µL, 12.4 mmol) at 0°C. The resulting mixture was stirred at the same temperature for 3 h. Then the reaction mixture was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, CHCl3–EtOAc=9 : 1) to afford 16 (1.12 g, 64%, 2 steps) as a pale yellow solid.
16: IR (film, cm−1): 3115, 2978, 2943, 1734, 1630, 1595, 1560, 1508, 1481, 1425, 1395, 1370, 1331, 1275, 1260, 1171, 1144, 1074, 1020, 966, 957, 833, 596; 1H-NMR (CDCl3, 500 MHz) δ: 8.59 (d, J=13.6 Hz, 1H), 7.76 (d, J=8.50 Hz, 1H), 7.42 (d, J=13.6 Hz, 1H), 6.91 (d, J=8.50 Hz, 1H), 4.04 (s, 3H), 1.66 (s, 9H); 13C-NMR (CDCl3, 125 MHz) δ: 165.2, 164.1, 149.1, 137.7, 137.4, 135.5, 118.9, 114.2, 84.0, 54.2, 28.1; HR-MS (ESI-TOF): Calcd for C13H16N2O5Na [(M+Na)+] 303.0954. Found 303.0958.
(S)-5-Benzhydryl 1-tert-Butyl 3-((S)-1-(2-Methoxy-6-(methoxycarbonyl)pyridin-3-yl)-2-nitroethyl)-2-oxopentanedioate (21)To a stirred solution of 15 (5.40 g, 22.7 mmol) in 1,2-dimethoxyethane (DME) (230 mL) were added α-ketoester 7d43) (8.77 g, 23.8 mmol) and Ni–diamine complex 20 (576 mg, 1.14 mmol) at −10°C. The resulting mixture was stirred at the same temperature for 48 h. The mixture was diluted with n-hexane, filtered through a pad of SiO2 and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=2 : 1) to afford 21 (12.1 g, 88%, dr>25 : 1) as a colorless oil. The ee value was determined by chiral HPLC analysis.
21: HPLC: DAICEL CHIRALCEL OD-H, n-hexane–isopropylalcohol (IPA)=9 : 1, 1.0 mL/min, 254 nm, τmajor 25.1 min, τminor 29.2 min; [α]D25 +4.4 (c=1.0, CHCl3, 95% ee); IR (film, cm−1): 1724, 1584, 1555, 1460, 1371, 1267, 1167, 1022, 982, 760, 702; 1H-NMR (CDCl3, 500 MHz) δ: 7.61 (d, J=7.37 Hz, 1H), 7.45 (d, J=7.37 Hz, 1H), 7.20–7.38 (m, 10H), 6.82 (s, 1H), 4.92 (dd, J=13.0, 9.07 Hz, 1H), 4.78 (dd, J=13.0, 4.53 Hz, 1H), 4.31–4.25 (m, 1H), 4.02 (s, 3H), 4.01–3.95 (m, 1H), 3.93 (s, 3H), 2.95 (dd, J=17.0, 9.64 Hz, 1H), 2.73 (dd, J=17.0, 4.53 Hz, 1H), 1.43 (s, 9H); 13C-NMR (CDCl3, 125 MHz) δ: 193.8, 170.0, 165.0, 161.0, 159.3, 145.3, 140.0, 139.4, 139.3, 128.5, 128.4, 128.1, 128.0, 127.2, 126.9, 122.5, 118.7, 84.4, 77.9, 75.1, 53.8, 52.6, 42.9, 41.6, 35.0, 27.5; HR-MS (ESI-TOF): Calcd for C32H35N2O10 [(M+H)+] 607.2286. Found 607.2312.
Methyl 5-((3S,4S,5R)-5-(tert-Butoxycarbonyl)-4-(2-methoxy-2-oxoethyl)pyrrolidin-3-yl)-6-methoxy Picolinate (24)To a stirred solution of 21 (2.33 g, 3.84 mmol) in MeOH (80 mL) was added Raney nickel (6.0 g, purchased from Aldrich, washed with water and MeOH) at room temperature. The resulting mixture was stirred under hydrogen pressure (900 psi) at 75°C for 2 h. Then, the reaction mixture was filtered through a pad of Celite and concentrated under reduced pressure. The crude material including 23 was applied to the following reaction without further purification.
To a stirred solution of the crude material including 23 in MeOH (40 mL) was added 10% Pd/C (1.0 g) at room temperature. The resulting mixture was stirred under ordinary hydrogen pressure (balloon) at the same temperature for 1.5 h. The reaction mixture was filtered through a pad of Celite and concentrated under reduced pressure. The crude material including carboxylic acid 37 was applied to the following reaction without further purification.
To a stirred solution of the crude material including 37 in MeOH (40 mL) was added SOCl2 (0.28 mL, 3.8 mmol) at 0°C. The resulting mixture was stirred at room temperature for 20 h. Then the reaction mixture was diluted with toluene and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, CHCl3–MeOH=96 : 4) to afford 24 (1.07 g, 68%, 3 steps) as a yellow oil.
24: [α]D25 −89.5 (c=0.97, CHCl3); IR (film, cm−1): 1740, 1462, 1263, 1211, 1159; 1H-NMR (CDCl3, 500 MHz) δ: 7.68 (br s, 2H), 4.69–4.63 (m, 1H), 4.15–4.08 (m, 2H), 4.07 (s, 3H), 3.96 (s, 3H), 3.89–3.82 (m, 1H), 3.74–3.66 (m, 1H), 3.34 (s, 3H), 2.36 (dd, J=8.00, 17.8 Hz, 1H), 2.18 (dd, J=5.75, 17.8 Hz, 1H), 1.46 (s, 9H); 13C-NMR (CDCl3, 125 MHz) δ: 170.8, 165.8, 165.3, 161.6, 144.7, 137.3, 122.3, 118.4, 85.6, 63.3, 54.1, 52.7, 51.7, 46.1, 40.6, 38.9, 30.5, 27.9; HR-MS (ESI-TOF): Calcd for C20H29N2O7 [(M+H)+] 409.1969. Found 409.1968.
(2R,3S,4S)-1-Benzyl 2-tert-Butyl 3-(2-Methoxy-2-oxoethyl)-4-(2-methoxy-6-(methoxycarbonyl)pyridin-3-yl)pyrrolidine-1,2-dicarboxylate (25)To a stirred solution of 24 (540 mg, 1.22 mmol) in CH2Cl2 (5 mL) were added Et3N (0.51 mL, 3.66 mmol) and CbzCl (0.26 mL, 1.83 mmol) at 0°C. The resulting mixture was stirred at room temperature for 5 h. Then the reaction was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=2 : 1) to afford 25 (589 mg, 89%) as a colorless amorphous solid. 25 exists as a mixture of rotamers in CDCl3 at 25°C.
25: [α]D25 −63.2 (c=0.90, CHCl3); IR (film, cm−1): 1742, 1721, 1709, 1460, 1412, 1368, 1287, 1265, 1213, 1155; 1H-NMR (CDCl3, 500 MHz) δ: 7.86 (d, J=7.37 Hz, 0.55H), 7.79 (d, J=7.37 Hz, 0.45H), 7.70–7.66 (m, 1H), 7.40–7.25 (m, 5H), 5.26–5.10 (m, 2H), 4.55 (d, J=8.50 Hz, 0.45H), 4.51 (d, J=8.50 Hz, 0.55H), 4.03 (s, 3H), 3.97–3.86 (m, 3H), 3.95 (s, 3H), 3.55 (s, 1.65H), 3.52 (s, 1.35H), 3.59–3.49 (m, 1H), 2.35 (dd, J=18.0, 6.24 Hz, 0.55H), 2.24 (dd, J=17.6, 6.80 Hz, 0.45H), 2.18 (dd, J=17.6, 8.50 Hz, 0.45H), 2.09 (dd, J=18.0, 8.50 Hz, 0.55H), 1.37 (s, 4H), 1.21 (s, 5H); 13C-NMR (CDCl3, 125 MHz) δ: 117.6, 171.5, 169.1, 168.9, 165.4, 161.4, 154.5, 154.4, 143.7, 143.6, 138.5, 138.1, 136.3, 136.1, 128.4, 128.3, 127.9, 127.8, 125.8, 125.5, 118.5, 118.4, 82.1, 82.0, 67.2, 67.1, 62.3, 61.5, 53.7, 52.5, 51.4, 50.5, 49.5, 40.5, 39.2, 39.0, 37.8, 31.2, 31.1, 27.7, 27.5; HR-MS (ESI-TOF): Calcd for C28H35N2O9 [(M+H)+] 543.2337. Found 543.2350.
(2S,3S,4S)-1-((Benzyloxy)carbonyl)-3-(2-methoxy-2-oxoethyl)-4-(2-methoxy-6-(methoxycarbonyl)pyridin-3-yl)pyrrolidine-2-carboxylic Acid (27)To a stirred solution of 25 (589 mg, 1.09 mmol) in CH2Cl2 (7.5 mL) was added TFA (2.5 mL) at room temperature. The resulting mixture was stirring at the same temperature for 19 h. Then the reaction mixture was concentrated under reduced pressure. The crude material including carboxylic acid 38 was applied to the following reaction without further purification.
To a suspension of crude material including 38 in Ac2O (5.5 mL) was added NaOAc (894 mg, 10.9 mmol) at room temperature. The resulting mixture was stirred at 110°C for 25 h. Then the mixture was concentrated under reduced pressure and the residue was diluted with water. After being stirred for 1 h, the mixture was extracted with CHCl3. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, CHCl3–MeOH=98 : 2) to afford 27 (370 mg, 70% 2 steps) as a colorless amorphous solid.
27: [α]D25 −55.3 (c=0.87, CHCl3); IR (film, cm−1): 1738, 1591, 1462, 1433, 1362, 1267, 1207, 1171, 1132; 1H-NMR (CDCl3, 500 MHz) δ: 10.08 (br s, 1H), 7.68 (d, J=7.37 Hz, 0.45H), 7.64 (d, J=7.37 Hz, 0.55H), 7.45–7.25 (m, 6H), 5.28–5.13 (m, 2H), 4.26 (d, J=5.10 Hz, 0.55H), 4.22 (d, J=5.10 Hz, 0.45H), 3.80–4.05 (m, 3H), 3.99 (s, 3H), 3.96 (s, 3H), 3.59 (s, 1.35H), 3.58 (s, 1.65H), 3.44–3.36 (m, 1H), 2.35–2.20 (m, 1H), 2.05–1.95 (m, 1H); 13C-NMR (CDCl3, 125 MHz) δ: 176.0, 175.1, 171.6, 171.5, 165.4, 161.4, 155.1, 154.3, 144.1, 136.6, 135.9, 128.5, 128.4, 128.2, 128.0, 127.7, 125.1, 125.0, 118.8, 118.6, 67.7, 67.6, 63.5, 63.0, 53.9, 52.6, 51.8, 49.0, 48.9, 42.8, 41.6, 39.2, 38.4, 33.1; HR-MS (ESI-TOF): Calcd for C24H25N2O9 [(M−H)–] 485.1555. Found 485.1554.
Acromelic Acid A (1)To a stirred solution of 27 (271 mg, 557 µmol) in H2O (1.5 mL) was added 5 M HBr in AcOH (6.0 mL) at room temperature. The resulting mixture was stirred at 100°C for 12 h. Then the reaction mixture was concentrated under reduced pressure. Purification of 1 was carried out according to the reported procedure.4) The residue was charged onto a column containing Dowex-50 WX8 hydrogen form (200–400 mesh). After elution with H2O (25 mL) and 3% aqueous NH3 (25 mL), the collected fractions were concentrated under reduced pressure. The resulting ammonium was charged onto a column containing Amberlite IRC-50 hydrogen form. After elution with H2O, the collected fractions were concentrated under reduced pressure to give free amino acid 1 (172 mg, quant) as a colorless solid.
1: mp >310°C (decomp.); [α]D25 +30.0 (c=1.11, H2O) [lit. [α]D +27.8 (c=0.35, H2O)]4); IR (film, cm−1): 3422, 1618, 1381, 787; 1H-NMR (D2O, 500 MHz) δ: 7.52 (d, J=7.37 Hz, 1H), 6.94 (d, J=7.37 Hz, 1H), 4.12 (d, J=7.37 Hz, 1H), 3.84–3.68 (m, 3H), 3.20–3.12 (m, 1H), 2.61 (dd, J=16.7, 5.10 Hz, 1H), 2.15 (dd, J=16.7, 10.2 Hz, 1H); 13C-NMR (D2O, 125 MHz) δ: 176.7, 173.6, 166.3, 163.1, 142.7, 139.5, 129.8, 108.9, 65.8, 47.5, 42.5, 42.4, 35.7; HR-MS (ESI-TOF): Calcd for C13H13N2O7 [(M−H)−] 309.0717. Found 309.0715.
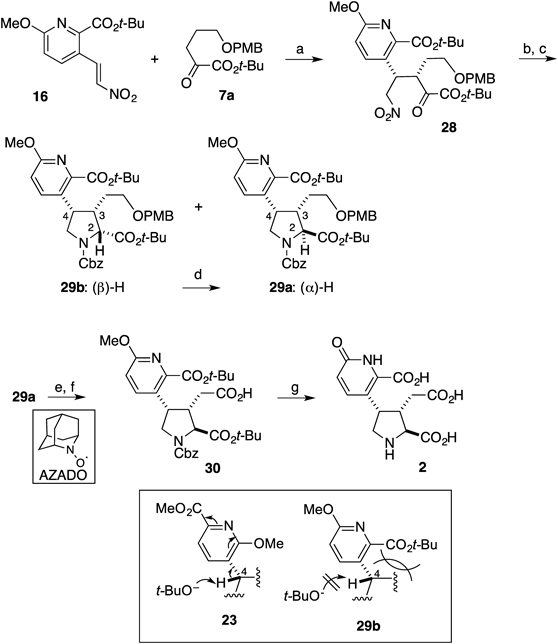
tert-Butyl 3-((2S,3S)-5-(tert-Butoxy)-3-(2-((4-methoxybenzyl)oxy)ethyl)-1-nitro-4,5-dioxopentan-2-yl)-6-methoxypicolinate (28)To a stirred solution of 16 (400 mg, 1.43 mmol) in IPA (0.4 mL) were added Ni–diamine complex 20 (36.2 mg, 71.4 µmol), α-ketoester 7a40,43) (880 mg, 2.85 mmol) in IPA (1.1 mL) and Et3N (49 mL, 357 mmol) at −10°C. The resulting mixture was stirred at the same temperature for 14 h. Then the solvent was removed under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=7 : 3) to afford 28 (887 mg, quant.) as a colorless oil. The ee value was determined by chiral HPLC analysis.
28: HPLC (DAICEL CHIRALPAK AD-H, n-hexane–IPA=19 : 1, 1.0 mL/min, 254 nm, τminor 14.1 min, tmajor 15.8 min); [α]D25 −33.0 (c=1.01, CHCl3, 91% ee); IR (film, cm−1): 2980, 2938, 2868, 1719, 1601, 1555, 1512, 1481, 1370, 1329, 1283, 1250, 1171, 1148, 1098, 1030, 847, 826; 1H-NMR (CDCl3, 500 MHz) δ: 7.57 (d, J=8.50 Hz, 1H), 7.17 (d, J=8.50 Hz, 2H), 6.85 (d, J=8.50 Hz, 2H), 6.74 (d, J=8.50 Hz, 1H), 4.87 (dd, J=13.0, 5.10 Hz, 1H), 4.81 (dd, J=13.0, 6.80 Hz, 1H), 4.47–4.38 (m, 1H), 4.30 (d, J=11.9 Hz, 1H), 4.26 (d, J=11.9 Hz, 1H), 4.09 (dt, J=9.6, 4.0 Hz, 1H), 3.92 (s, 3H), 3.79 (s, 3H), 3.49–3.41 (m, 2H), 2.18–2.00 (m, 2H), 1.64 (s, 9H), 1.36 (s, 9H); 13C-NMR (CDCl3, 125 MHz) δ: 195.0, 165.3, 162.4, 159.8, 159.2, 147.3, 139.2, 129.7, 129.3, 125.3, 113.7, 113.0, 83.7, 82.7, 77.6, 72.3, 67.3, 55.3, 53.5, 46.0, 39.9, 31.1, 28.1, 27.6; HR-MS (ESI-TOF): Calcd for C30H40N2O10Na [(M+Na)+] 611.2575. Found 611.2570.
1-Benzyl 2-(tert-Butyl) (2S,3S,4S)-4-(2-(tert-Butoxycarbonyl)-6-methoxypyridin-3-yl)-3-(2-((4-methoxybenzyl)oxy)ethyl)pyrrolidine-1,2-dicarboxylate (29a) and 1-Benzyl 2-(tert-Butyl) (3S,4S)-4-(2-(tert-Butoxycarbonyl)-6-methoxypyridin-3-yl)-3-(2-((4-methoxybenzyl)oxy)ethyl)pyrrolidine-1,2-dicarboxylate (29b)To a stirred solution of 28 (7.77 g, 13.2 mmol) in MeOH (65 mL) was added Raney nickel (23 g, purchased from Aldrich, washed with water and MeOH) at room temperature. The resulting mixture was stirred under hydrogen pressure (700 psi) at room temperature for 1.5 h. The mixture was filtered through a pad of Celite and concentrated under reduced pressure. The crude materials including pyrrolidines 39a and b were applied to the following reaction without further purification.
To a solution of the crude materials including 39a and b in CH2Cl2 (70 mL) were added Et3N (3.66 mL, 26.4 mmol) and CbzCl (2.81 mL, 19.8 mmol) at 0°C. The resulting mixture was stirred at the same temperature for 30 min. Then the reaction mixture was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=3 : 1) to afford 29a (2.30 g, 26% 2steps) as a colorless oil and 29b (4.19 g, 47% 2 steps) as a colorless oil. These compounds exist as a mixture of rotamers in CDCl3 at 25°C.
29a: [α]D20 −17.2 (c=1.03, CHCl3); IR (film, cm−1): 2978, 2938, 2870, 1736, 1709, 1599, 1512, 1481, 1414, 1368, 1356, 1331, 1281, 1248, 1157, 1032, 824, 698; 1H-NMR (CDCl3, 500 MHz) δ: 7.44–7.28 (m, 6H), 7.21–7.13 (m, 2H), 6.86–6.70 (m, 3H), 5.22–5.07 (m, 2H), 4.36–4.33 (m, 2H), 4.26–4.21 (m, 1H), 4.11–4.04 (m, 1H), 3.96–3.85 (m, 4H), 3.80–3.62 (m, 4H), 3.48–3.19 (m, 2H), 2.80–2.70 (m, 1H), 1.70–1.33 (m, 20H); 13C-NMR (CDCl3, 125 MHz) δ: 171.2, 165.9, 162.0, 159.1, 154.8, 154.5, 148.2, 148.1, 138.5, 136.6, 136.4, 130.3, 129.2, 129.1, 128.5, 128.4, 128.0, 127.9, 127.8, 127.7, 124.9, 113.7, 112.4, 82.5, 82.4, 81.7, 81.6, 72.7, 68.3, 68.0, 67.1, 64.8, 64.4, 55.2, 53.5, 50.0, 45.0, 43.6, 40.4, 39.5, 29.1, 29.0, 28.1, 28.0, 27.8; HR-MS (ESI-TOF) Calcd for C38H48N2O9Na [(M+Na)+] 699.3252. Found 699.3263.
29b: [α]D20 −31.0 (c=1.18, CHCl3); IR (film, cm−1): 3002, 2977, 2941, 2904, 1740, 1721, 1709, 1698, 1601, 1513, 1480, 1412, 1368, 1329, 1281, 1248, 1169, 1144, 1032, 847, 824, 755, 698; 1H-NMR (CDCl3, 500 MHz) δ: 7.89 (t, J=8.50 Hz, 1H), 7.40–7.27 (m, 5H), 7.21–7.08 (m, 2H), 6.85–6.72 (m, 3H), 5.22–5.08 (m, 2H), 4.49 (d, J=9.1 Hz, 0.45H), 4.44 (d, J=9.1 Hz, 0.55H), 4.36–4.20 (m, 2H), 4.14–3.89 (m, 2H), 3.93 (s, 3H), 3.83–3.70 (m, 1H), 3.78 (s, 3H), 3.40–3.00 (m, 3H), 1.66–1.54 (m, 11H), 1.44 (s, 4H), 1.29 (s, 5H); 13C-NMR (CDCl3, 125 MHz) δ: 170.5, 170.4, 166.2, 162.0, 159.1, 154.8, 154.4, 148.1, 147.9, 140.2, 140.1, 137.9, 136.5, 136.3, 130.5, 129.2, 128.9, 128.5, 128.4, 128.0, 127.9, 126.4, 126.1, 113.7, 113.6, 112.6, 112.5, 82.6, 82.5, 82.2, 82.1, 72.5, 72.4, 68.3, 68.2, 67.2, 67.1, 63.4, 62.9, 55.2, 53.5, 52.4, 51.8, 41.9, 40.9, 40.8, 39.9, 28.2, 28.0, 27.8, 27.7, 27.6; HR-MS (ESI-TOF): Calcd for C38H48N2O9Na [(M+Na)+] 699.3252. Found 699.3246.
Epimerization of 29b to aTo a stirred solution of 29b (60 mg, 0.884 µmol) in the 9 : 1 mixture of t-BuOH and benzene (total 2.0 mL) was added t-BuOK (14.9 mg, 0.133 µmol) at 0°C. The resulting mixture was stirred at room temperature for 6 h. Then the reaction mixture was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=3 : 1) to afford 29a (37 mg, 62%) as a colorless oil.
1-Benzyl 2-(tert-Butyl) (2S,3S,4S)-4-(2-(tert-Butoxycarbonyl)-6-methoxypyridin-3-yl)-3-(2-hydroxyethyl)pyrrolidine-1,2-dicarboxylate (40)To a stirred solution of 29a (100 mg, 147 mmol) in the 20 : 1 mixture of CH2Cl2 and H2O (0.735 mL) was added 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) (50 mg, 0.221 mmol) at room temperature. The resulting mixture was stirred at the same temperature for 1 h. Then the reaction mixture was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=1 : 1 and n-hexane–Et2O=1 : 2) to afford alcohol 40 (89.9 mg, 99%) as a colorless oil. This compound exists as a mixture of rotamers in CDCl3 at 25°C.
40: [α]D25 −34.0 (c=1.45, CHCl3); IR (film, cm−1): 2978, 2936, 1740, 1719, 1701, 1690, 1655, 1597, 1560, 1481, 1458, 1420, 1368, 1331, 1283, 1157, 1028, 847, 698; 1H-NMR (CDCl3, 500 MHz) δ: 7.43–7.28 (m, 6H), 6.77 (d, J=8.50 Hz, 0.55H), 6.74 (d, J=8.50 Hz, 0.45H), 5.21–5.11 (m, 2H), 4.18 (d, J=5.10 Hz, 0.45H), 4.16 (d, J=5.10 Hz, 0.55H), 4.09–4.01 (m, 1H), 3.93 (s, 3H), 3.92–3.86 (m, 1H), 3.80–3.67 (m, 1H), 3.64–3.54 (m, 2H), 2.81–2.72 (m, 1H), 1.70–1.35 (m, 2H), 1.59 (s, 9H), 1.49 (s, 4H), 1.39 (s, 5H); 13C-NMR (CDCl3, 125 MHz) δ: 171.5, 171.4, 166.2, 162.1, 154.7, 154.4, 148.1, 148.0, 138.3, 136.5, 136.3, 128.5, 128.4, 128.0, 127.9, 125.5, 125.4, 112.7, 82.8, 82.7, 82.0, 67.3, 67.2, 64.8, 64.4, 60.9, 60.8, 53.5, 50.6, 44.9, 43.8, 40.5, 39.6, 32.1, 28.1, 27.9, 27.8; HR-MS (ESI-TOF) Calcd for C30H41N2O8 [(M+H)+] 557.2857. Found 557.2858.
2-((2R,3S,4S)-1-((Benzyloxy)carbonyl)-2-(tert-butoxycarbonyl)-4-(2-(tert-butoxycarbonyl)-6-methoxypyridin-3-yl)pyrrolidin-3-yl)acetic Acid (30)To a stirred solution of 40 (250 mg, 449 mmol) in the 1 : 1 mixture of CH2Cl2–phosphate buffer (pH 7.6) (total 2.7 mL) were added AZADO (13.7 mg, 89.8 mmol) and PhI(OAc)2 (434 mg, 1.34 mmol) at 0°C. The resulting mixture was stirred at the same temperature for 8 h. Then the mixture was added to saturated aqueous Na2S2O3 at 0°C. After 1 h, the resulting mixture was extracted with EtOAc. The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, n-hexane–EtOAc=1 : 1) to afford 30 (222 mg, 87%) as a colorless amorphous solid. This compound exists as a mixture of rotamers in CDCl3 at 25°C.
30: [α]D25 −39.6 (c=1.05, CHCl3); IR (film, cm−1): 2980, 2941, 2906, 1710, 1599, 1560, 1481, 1413, 1367, 1332, 1282, 1253, 1228, 1161, 1093, 1030, 844, 736, 698; 1H-NMR (CDCl3, 500 MHz) δ: 7.42–7.28 (m, 6H), 6.77 (d, J=8.50 Hz, 0.55H), 6.74 (d, J=8.50 Hz, 0.45H), 5.22–5.08 (m, 2H), 4.20–4.11 (m, 2H), 3.96–3.89 (m, 1H), 3.92 (s, 3H), 3.80–3.67 (m, 1H), 3.21–3.11 (m, 1H), 2.27–2.16 (m, 2H), 1.57 (s, 9H), 1.47 (s, 4H), 1.37 (s, 5H); 13C-NMR (CDCl3, 125 MHz) δ: 176.4, 176.3, 170.4, 165.6, 162.2, 154.6, 154.4, 148.1, 138.0, 136.4, 136.2, 128.4, 128.3, 128.0, 127.9, 127.8, 124.6, 124.5, 112.8, 82.7, 82.6, 82.0, 81.9, 67.3, 64.9, 64.6, 53.5, 50.1, 49.9, 43.6, 42.6, 39.9, 39.0, 33.8, 28.0, 27.9, 27.7; HR-MS (ESI-TOF): Calcd for C30H38N2O9Na [(M+Na)+] 593.2470. Found 593.2486.
Acromelic Acid B (2)To a stirred solution of 30 (195 mg, 342 µmol) in H2O (1.7 mL) was added 30% HBr in AcOH (3.4 mL) at room temperature. The resulting mixture was stirred at 100°C for 36 h. Then the reaction mixture was concentrated under reduced pressure. The residue was charged onto a column containing Dowex-50 WX8 hydrogen form (200–400 mesh). After elution with H2O and 3% aqueous NH3, the collected fractions were concentrated under reduced pressure. The resulting ammonium salt was charged onto a column containing Amberlite IRC-50 hydrogen form. After elution with H2O, the collected fractions were concentrated under reduced pressure to give free amino acid 2 (105 mg, 99%) as a colorless amorphous solid.
2: [α]D20 −68.8 (c=0.98, H2O) [lit. [α]D27 −74.0 (c=0.1, H2O)]37); IR (film, cm−1) 3300–2700, 1655, 1597, 1419, 1363, 1251, 1167, 1060, 842, 801, 673. 1H-NMR (D2O, 500 MHz) δ: 7.68 (d, J=9.16 Hz, 1H), 6.70 (d, J=9.16 Hz, 1H), 4.65 (dt, J=11.5, 8.0 Hz, 1H), 4.08 (d, J=5.7 Hz, 1H), 3.80 (dd, J=11.5, 8.0 Hz, 1H), 3.65 (t, J=11.5 Hz, 1H), 3.27–3.20 (m, 1H), 2.52 (dd, J=16.6, 6.3 Hz, 1H), 2.35 (dd, J=16.6, 8.6 Hz, 1H); 13C-NMR (D2O, 125 MHz) δ: 175.7, 173.0, 166.5, 163.1, 143.3, 141.2, 120.4, 115.3, 65.5, 47.3, 42.3, 38.6, 34.7; HR-MS (ESI-TOF): Calcd for C13H15N2O7 [(M+H)+] 311.0874. Found 311.0877.