Results and Discussion
First Approach to the Synthesis of 1: Introduction of a Cysteine Unit via an Epoxide Ring-Opening ReactionInitially, we hypothesized that the unknown absolute configuration of 1 would be similar to the configuration shown in Fig. 1, which is based on the absolute configuration of decaline units found in several natural labdane-type diterpenes8) and our previous report4) on the relative configuration. For the synthesis of the reported cyslabdan structure (1), the introduction of the N-acetyl-L-cysteine unit at the sterically hindered C17 (cyslabdan numbering), in which the neopentyl-like position is axial, was recognized as the most challenging step. To complete this step, the selection and introduction of a suitable leaving group at C17 to allow an SN2 reaction is necessary. This also requires the protection of the sterically hindered C8-tertiary hydroxyl group.9) Taking these into consideration, the initial synthetic strategy for the reported structure of cyslabdan (1) was designed as shown in Chart 1, in which epoxide 3 was adopted as a key intermediate. Although the protection of the sterically hindered C8-tertiary hydroxy group in this strategy was unnecessary, the requisite epoxide ring-opening reaction with N-acetyl-L-cysteine was anticipated to be difficult given that the direction of nucleophilic attack on the epoxide was very sterically hindered and fixed. However, we expected this synthetic strategy to afford a concise and short route to the reported structure of cyslabdan (1). The diene unit of the epoxide 3 would be constructed from diol 4, which could be synthesized via the stereoselective epoxidation and configurational inversion of the C7-hydroxy group of the known allylic alcohol 5, which is readily prepared from commercially available (3aR)-(+)-sclareolide.10)
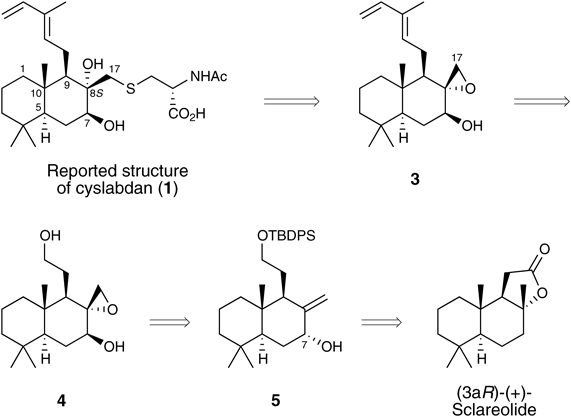
Chart 1. Retrosynthetic Analysis of 1
Our synthesis commenced with the stereoselective epoxidation of the known allylic alcohol 5 with m-chloroperbenzoic acid (mCPBA) to give 6 (Chart 2). The stereochemistry of epoxide 6 was determined by the conversion of 6 to the known triol 711) and the comparison of their physicochemical properties. The Mitsunobu reaction12) of 6 led primarily to dehydration; however, configurational inversion at the C7-hydroxy group was achieved via Dess–Martin oxidation13,14) and reduction with LiAl(t-BuO)3H to give 8. Desilylation of 8 with tetrabutylammonium fluoride (TBAF) gave diol 4, which was subjected to double protection as a bis-triethylsilyl (TES) ether followed by Swern oxidation15) to afford aldehyde 9. Stereoselective diene construction via a subsequent four-step sequence involving standard methodologies including Wittig olefination gave 10, which was exposed to TBAF to afford the desired epoxide 3. The stereochemistry at each chiral center in 3 was confirmed using nuclear Overhauser effect (NOE) experiments.
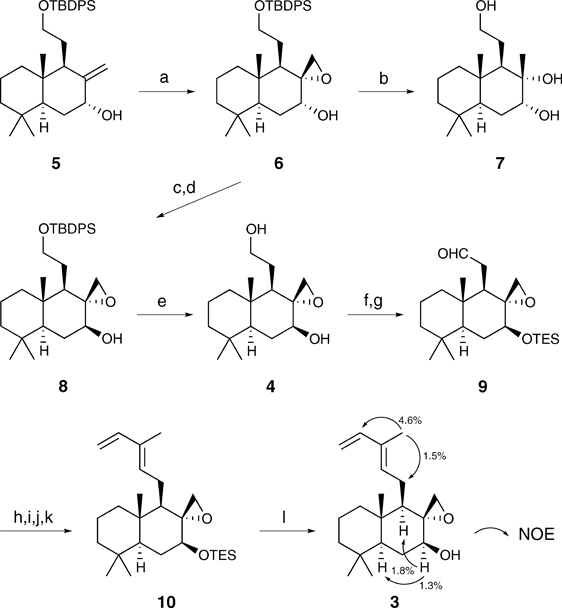
Chart 2. Synthesis of the Key Intermediate Epoxide
3Reagents and conditions: (a) mCPBA, CH2Cl2, 0°C, 94%; (b) LAH, Et2O, r.t., 91%; (c) DMP, CH2Cl2, r.t.; (d) LiAl(t-BuO)3H, Et2O, r.t., 94% (2 steps); (e) TBAF, THF, r.t., 99%; (f) TESCl, DMAP, imidazole, DMF, 0°C, 93%; (g) (COCl)2, DMSO, Et3N, CH2Cl2, −78°C, 99%; (h) Ph3P=C(Me)CO2Et, benzene, 60°C, 96%; (i) DIBAL, CH2Cl2, −78°C; (j) MnO2, CH2Cl2, r.t.; (k) Ph3PMeBr, NaHMDS, THF, 0°C, 74% (3 steps); (l) TBAF, THF, r.t., 91%.
With the epoxide 3 in hand, the epoxide ring-opening reaction was attempted using various nucleophiles (Table 1). The treatment of 3 with N-acetyl-L-cysteine t-butyl ester in the presence of t-BuOK in t-BuOH at 40°C led to no reaction (run 1). Therefore, thiol, thiolate ions, and iodide ions were investigated as nucleophiles under harsh conditions (runs 2–6); these reactions also did not proceed. Subsequent epoxide ring-opening reactions in the presence of MgBr2·Et2O as a Lewis acid (run 7) and under acidic conditions (run 8) were similarly unsuccessful. These results were attributed to the steric hindrance at the site of nucleophilic attack and forced us to explore an alternative synthetic route.
Table 1. Attempted Nucleophilic Ring-Opening of Epoxide
3 with Various Nucleophiles
 |
|---|
| Run | Reagents (equiv.) | Solvent(s) | Temp. (°C) | Time (h) | Result |
|---|
| 1 | N-Acetyl-L-cysteine t-butyl ester (5), t-BuOK (2.5) | t-BuOH | 40 | 24 | No reaction |
| 2 | NaSH (2) | MeOH | 50 | 48 | No reaction (R=SH) |
| 3 | NaSH (10), 15-crown-5 (12) | (CH2OH)2–DMF | 50 | 96 | No reaction (R=SH) |
| 4 | EtSH (3), NaH (3) | DMF | r.t. | 16 | No reaction (R=SEt) |
| 5 | KSAc (8) | DMF | 50 | 48 | No reaction (R=SAc) |
| 6 | NaI (10) | DMF | 140 | 24 | No reaction (R=I) |
| 7 | TBAI (5), MgBr2·Et2O (2) | DMF | 60 | 23 | Decomposed |
| 8 | HClO4 (5) | THF–H2O | 60 | 16 | No reaction (R=OH) |
The second synthetic strategy for 1 is shown in Chart 3. The key steps in this synthesis are the SN2 reaction of substrate 13 bearing a triflate-leaving group at the sterically hindered C17 position with an N-acetyl-L-cysteine methyl ester and the subsequent steric inversion of the C7-hydroxy group in the presence of a variety of other functional groups. In the SN2 reaction, the sterically hindered C8-tertiary hydroxyl group in 13 must be protected. Therefore, a cyclic carbonate protecting group was selected to protect both the sterically hindered C8-tertiary hydroxyl group and the C7-hydroxy group. The triflate 13 including diene and C8-17 diol units was envisaged to be stereoselectively prepared from benzyl ether 14 via Wittig reaction and dihydroxylation. The benzyl ether 14 can be derived from commercially available (3aR)-(+)-sclareolide.
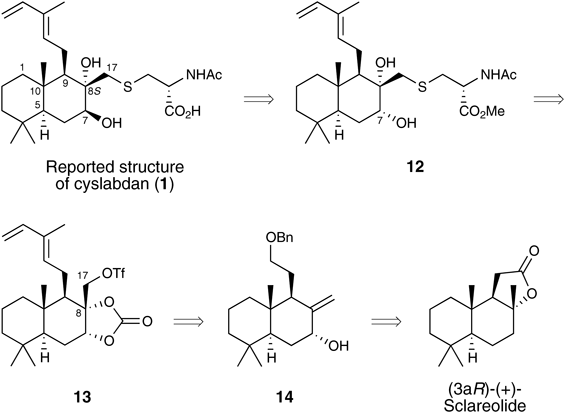
Chart 3. Second Retrosynthetic Analysis of 1
The benzyl ether 14 was prepared in four steps from (3aR)-(+)-sclareolide in a manner similar to that described for the synthesis of 510) (Chart 4). The dihydroxylation of 14 with OsO4 afforded triol 16 stereoselectively, which was then subjected to the selective protection of the primary hydroxy group as a tert-butyldimethylsilyl (TBDPS) ether followed by the formation of the cyclic carbonate via treatment with triphosgene to give 17. Diene 18 was then constructed via a four-step sequence involving the deprotection of the benzyl ether, Dess–Martin oxidation, and Wittig reactions with Ph3P=C(Me)CHO and Ph3PMeBr. Exposure of 18 to TBAF provided alcohol 19. The stereochemistry of each chiral center in 19 was confirmed using rotating-frame Overhauser effect spectroscopy (ROESY) experiments.
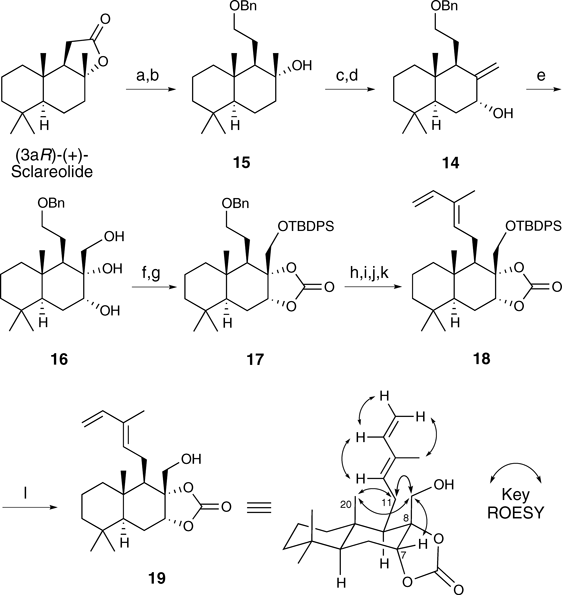
Chart 4. Synthesis of the Key Intermediate
19Reagents and conditions: (a) LAH, THF, 0°C; (b) BnCl, NaH, TBAI, DMF, 0°C to r.t., 93% (2 steps); (c) SOCl2, DMAP, pyridine, −30°C; (d) SeO2, TBHP, salicylic acid, CH2Cl2, r.t., 55% (2 steps); (e) OsO4, NMO, THF–H2O, r.t., 69%; (f) TBDPSCl, imidazole, DMAP, CH2Cl2, r.t., 95%; (g) triphosgene, pyridine, CH2Cl2, 0°C, 92%; (h) H2, Pd/C, MeOH–THF, r.t.; (i) DMP, CH2Cl2, 0°C to r.t., 96% (2 steps); (j) Ph3P=C(Me)CHO, toluene, reflux, 69%; (k) Ph3PMeBr, n-BuLi, THF, −78 to 0°C, 87%; (l) TBAF, AcOH, THF, 0°C to r.t., 99%.
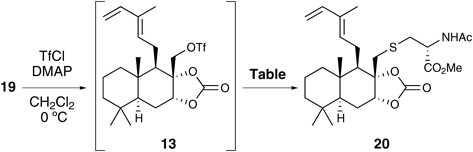
Next, the SN2 reaction of N-acetyl-L-cysteine methyl ester at the sterically hindered C17 position of triflate 13 was investigated (Table 2). Alcohol 19 was converted to the triflate 13 by treating it with trifluoromethanesulfonyl chloride, which was then subjected to the SN2 reaction without purification due to its instability. The treatment of 13 with N-acetyl-L-cysteine methyl ester in pyridine at room temperature led to no reaction. When the reaction mixture was gradually warmed to 90°C, 13 decomposed without giving the desired product 20 (run 1). Subsequently, the SN2 reaction with the sodium thiolate derivative of N-acetyl-L-cysteine methyl ester (prepared by pretreatment with sodium hydride) in tetrahydrofuran (THF) succeeded in affording the desired product 20 in 61% yield (run 2). Exchanging THF with N,N-dimethylformamide (DMF) as the reaction solvent increased the yield of the desired product to 76% (run 3).
Table 2.
SN2 Reactions of Triflate
13 with
N-Acetyl-L-cysteine Methyl Ester
 |
|---|
| Run | Reagents (eq.) | Solvent | Temp. | Time | Result (%) |
|---|
| 1 | N-Acetyl-L-cysteine methyl ester (3) | Pyridine | r.t. to 90°C | 16 h | Decomposed |
| 2 | N-Acetyl-L-cysteine methyl ester (3), NaH (3.1) | THF | 0°C to r.t. | 1 h | 20 (61%) |
| 3 | N-Acetyl-L-cysteine methyl ester (3), NaH (2.5) | DMF | 0°C | 5 min | 20 (76%) |
The final conversion of 20 to the reported structure of cyslabdan (1) was then investigated (Chart 5). Carbonate 20 was hydrolyzed under alkali conditions to give acid 12, which was subjected to Parikh–Doering oxidation16) followed by reduction with NaBH4 to afford 1 in low overall yield.17) Notably, although the synthesis of the reported structure of cyslabdan (1) was achieved, the 1H-NMR spectrum of the synthetic material did not coincide with the reported data for natural cyslabdan.4)

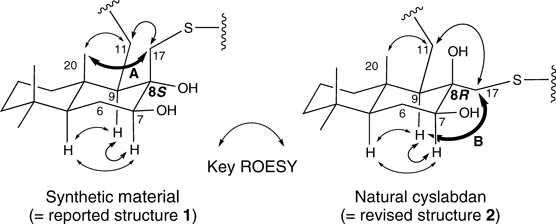
Therefore, natural cyslabdan was newly isolated from the culture broth of actinomycete strain K04-0144,18) and ROESY NMR analyses of both the synthetic material (reported structure 1) and natural cyslabdan were performed. A key cross signal A between the C20–CH3 and C17–CH2 protons that was observed for the synthetic material was not detected for the natural cyslabdan; instead, a different cross signal B between the C9–CH and C17–CH2 protons was observed for the natural compound (Fig. 2). These results suggest that the stereochemistry of C8 was 8R not 8S. We carefully reviewed the reported data in 2008 to determine the absolute configuration of natural cyslabdan and understood that our previous report, in which we had determined that the stereochemistry of C8 was 8S despite the lack of the key cross signal A and the existence of the key cross signal B, was incorrect.
Synthesis of Cyslabdan (Revised Structure 2) and Determination of Its Absolute ConfigurationIn light of our past research results, the synthesis of (8R)-cyslabdan (2) was attempted. The retrosynthetic analysis of 2 is shown in Chart 6. The connection of the decalin and N-acetyl-L-cysteine components was envisioned to occur via an SN2 reaction between epoxide 21 and N-acetyl-L-cysteine. In contrast to the synthesis of 1, this SN2 reaction was expected to proceed without any difficulty. The epoxide 21 would be synthesized from lactol 22 through the formation of a diene and an epoxide. Lactol 22 could be derived from allylic alcohol 23 via the stereoinversion of the C7-hydroxy group and stereoselective iodolactonization. Compound 23 was expected to be prepared from commercially available (3aR)-(+)-sclareolide.
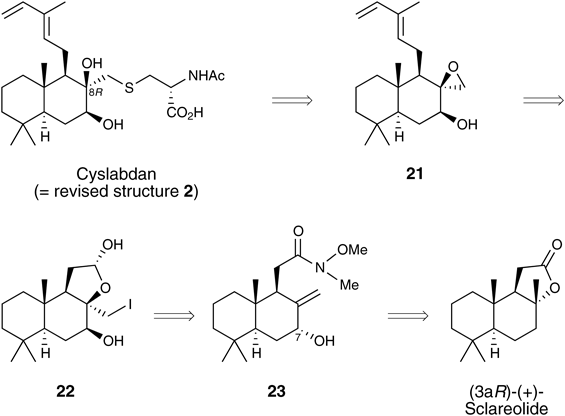
Chart 6. Retrosynthetic Analysis of 2
First, (3aR)-(+)-sclareolide was treated with N,O-dimethylhydroxylamine in the presence of trimethylaluminum to afford Weinreb amide 2419) (Chart 7). Dehydration of 24 with thionyl chloride gave exo-olefin 25,20–31) which was subjected to allylic oxidation with SeO232) to provide allylic alcohol 23. The subsequent 5-exo-tet iodolactonization of 23 with N-iodosuccinimide (NIS) proceeded smoothly and stereoselectively to afford the desired iodolactone 26. The pyridinium chlorochromate (PCC) oxidation of 26 provided ketone 27, which was reduced stereoselectively with diisobutylaluminium hydride (DIBAL) to give 22 and other reduction products.33) The stereochemistry of each chiral center in 22 was confirmed using ROESY experiments.

Next, we investigated the diene construction and the connection of the decalin unit with N-acetyl-L-cysteine (Chart 8). The treatment of lactol 22 with Ph3P=C(Me)CHO gave the desired aldehyde 28 via one-pot epoxide formation and stereoselective olefination; however, the yield was very low due to the generation of decomposition products. Therefore, we attempted the stepwise synthesis of 28. The Wittig reaction of 22 with Ph3P=CH2 afforded olefin 29 in high yield, which was subjected to cross metathesis with methacrolein in the presence of Grubbs second-generation catalyst34–38) to afford the desired aldehyde 28, which was also subjected to Wittig reaction with Ph3P=CH2 to give epoxydiene 21. Finally, the coupling of epoxydiene 21 with N-acetyl-L-cysteine under mild basic conditions afforded (8R)-cyslabdan (2). As expected, the synthetic (8R)-cyslabdan (2) was completely identical to an authentic sample of cyslabdan in all respects (1H- and 13C-NMR, ROESY correlations, [α]D, IR, MS, ultrafast liquid chromatography (UFLC), and retention time). Especially, the similarity in the optical rotations ([α]D26=+32.4, c=0.1, MeOH for synthetic 2 and [α]D25=+26.8, c=0.1, MeOH for natural cyslabdan4)) established the absolute configuration of natural cyslabdan as being identical to that of 2.

Experimental
GeneralAll reactions were conducted in flame-dried glassware under a nitrogen atmosphere employing standard techniques for the handling of air-sensitive materials. Commercial reagents were used without further purification unless otherwise indicated. Organic solvents were distilled and dried over 3 or 4 Å molecular sieves. Cold baths were prepared using ice/water (0°C) and dry ice/acetone (−78°C). Purification by flash column chromatography was performed over silica gel 60 N (spherical, neutral, particle size 40–50 µm). TLC was performed on 0.25 mm Merck silica gel 60 F254 plates and visualized using UV light (254 nm) along with phosphomolybdic acid and p-anisaldehyde TLC stains. Unless otherwise noted, yields are reported on chromatographically and spectroscopically pure compounds. 1H-, 13C-NMR spectra were recorded using an internal deuterium lock on a JNM-EX270 instrument (JEOL, Tokyo, Japan) or 400-MR, VNMRS-400, and UNITY-400 spectrometers (Agilent Technologies, Waldbronn, Germany). All NMR signals are reported in ppm relative to the internal reference standard provided by chloroform (i.e., 7.26, 77.0 ppm for the 1H-, 13C-NMR spectra, respectively). Multiplicity data are presented as follows: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, br=broad, dd=doublet of doublets, dt=doublet of triplets, and ddd=doublet of doublet of doublets. Coupling constants (J) are reported in Hz. IR spectra were recorded on an FT/IR460-plus IR spectrometer (JASCO, Tokyo, Japan), and absorption data are provided in wavenumbers (cm−1). Optical rotations were recorded on a DIP-1000 polarimeter (JASCO) and reported as follows: [α]TD, concentration (g/100 mL), and solvent. High-resolution (HR)-MS were obtained on a JMS-700 Mstation, JEOL JMS-AX505HA or JEOL JMS-T100LP system (JEOL) equipped with an FAB, electron ionization (EI), or electrospray ionization (ESI) HR-MS, respectively.
2-((1R,2R,4aS,8aS)-2-Hydroxy-2,5,5,8a-tetramethyldecahydronaphthalen-1-yl)-N-methoxy-N-methylacetamide (24)A solution of MeO(Me)NH·HCl (390 mg, 3.99 mmol) in CH2Cl2 (2.0 mL) was treated with Me3Al (1.06 M in n-hexane, 3.90 mL, 4.19 mmol) at 0°C. After stirring for 2 h at 0°C, a solution of (3aR)-(+)-sclareolide (500 mg, 2.00 mmol) in CH2Cl2 (2.0 mL) was added. After stirring for an additional 2 h at 0°C, the reaction mixture was quenched with 10% aq H2SO4. The aqueous layer was extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (1 : 2 hexanes/EtOAc) to afford 24 (571 mg, 92%) as a colorless amorphous solid. The spectroscopic data for 24 were identical to those reported in the literature.31)
N-Methoxy-N-methyl-2-((1S,4aS,8aS)-5,5,8a-trimethyl-2-methylenedecahydronaphthalen-1-yl)acetamide (25)To a solution of 24 (10.0 g, 32.1 mmol) and pyridine (4.7 mL, 64.2 mmol) in CH2Cl2 (200 mL) was added dropwise a solution of SOCl2 (11.7 mL, 161 mmol) and pyridine (23.3 mL, 289 mmol) in CH2Cl2 (90 mL) at −78°C. After stirring for 0.5 h at −78°C, the reaction mixture was quenched with a saturated aq Na2CO3 solution. The aqueous layer was extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (4 : 1 hexanes/EtOAc) to afford 25 (8.55 g, 90%) as a colorless amorphous solid. The spectroscopic data for 25 were identical to those reported in the literature.31)
2-((1R,3R,4aS,8aS)-3-Hydroxy-5,5,8a-trimethyl-2-methylenedecahydronaphthalen-1-yl)-N-methoxy-N-methylacetamide (23)A solution of 25 (5.00 g, 17.0 mmol) in CH2Cl2 (85 mL) was treated with SeO2 (37.8 mg, 0.341 mmol), salicylic acid (235 mg, 1.70 mmol), and tert-butylhydroperoxide (TBHP) (5.0 M solution in decane, 10.2 mL, 51.1 mmol) at room temperature. After stirring for 24 h at room temperature, the reaction mixture was quenched with a saturated aq Na2S2O3 solution. The aqueous layer was extracted with CH2Cl2. The organic layer was combined, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (1 : 1 hexanes/EtOAc) to afford 23 (3.22 g, 61%) as an amorphous brown solid: [α]D26–69.1 (c=1.0, CHCl3); IR (neat) cm−1: 3455, 3021, 1648, 1388, 1215, 1036; 1H-NMR (400 MHz, CDCl3) δ: 4.91 (s, 1H), 4.52 (s, 1H), 4.31 (t, 1H, J=2.8 Hz), 3.68 (s, 3H), 3.10 (s, 3H), 2.93 (dd, 1H, J=10.4, 3.6 Hz), 2.60 (dd, 1H, J=16.0, 10.4 Hz), 2.39 (dd, 1H, J=16.0, 3.6 Hz), 1.85 (dt, 1H, J=13.2, 2.8 Hz), 1.75 (dd, 1H, J=13.2, 2.8 Hz), 1.55–1.13 (m, 7H), 0.86 (s, 3H), 0.77 (s, 3H), 0.67 (s, 3H); 13C-NMR (400 MHz, CDCl3) δ: 174.4, 150.8, 109.1, 73.5, 61.4, 47.2, 46.2, 42.0, 39.1, 38.7, 33.3, 33.1, 32.5, 30.5, 26.9, 21.7, 19.3, 13.8; HR-MS (ESI) [M+Na]+ Calcd for C18H31NNaO3 332.2202. Found 332.2203.
(3aR,4R,5aS,9aS,9bR)-4-Hydroxy-3a-(iodomethyl)-6,6,9a-trimethyldecahydronaphtho[2,1-b]furan-2(3aH)-one (26)To a solution of 23 (2.71 g, 8.75 mmol) in MeCN–H2O (2 : 1, 88 mL) was added NIS (3.94 g, 17.5 mmol) at room temperature. After stirring for 1 h at room temperature, the reaction mixture was quenched with a saturated aq Na2CO3 solution and a saturated aq Na2S2O3 solution. The aqueous layer was extracted with EtOAc. The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (4 : 1 hexanes/EtOAc) to afford 26 (3.17 g, 92%) as an amorphous yellow solid: [α]D26 –13.5 (c=1.0, CHCl3); IR (neat) cm−1: 3488, 2851, 2067, 1771, 1418, 1214, 1060; 1H-NMR (400 MHz, CDCl3) δ: 4.34 (t, 1H, J=8.8 Hz), 3.96 (d, 1H, J=11.2 Hz), 3.27 (d, 1H, J=11.2 Hz), 2.82 (dd, 1H, J=19.2, 11.2 Hz), 2.40 (dd, 1H, J=19.2, 4.0 Hz), 2.12 (dd, 1H, J=11.2, 4.0 Hz), 1.93 (dt, 1H, J=18.4, 8.8 Hz), 1.61–0.86 (m, 8H), 0.93 (s, 3H), 0.91 (s, 3H), 0.86 (s, 3H); 13C-NMR (400 MHz, CDCl3) δ: 176.2, 88.9, 70.1, 54.9, 48.2, 42.6, 41.6, 37.0, 33.6, 32.3, 31.1, 27.2, 21.2, 18.1, 17.0, 15.5; HR-MS (ESI) [M+Na]+ Calcd for C16H25NaIO3 415.0746. Found 415.0741.
(3aR,5aS,9aS,9bR)-3a-(Iodomethyl)-6,6,9a-trimethyloctahydronaphtho[2,1-b]furan-2,4(3aH,5H)-dione (27)A suspension of 26 (600 mg, 1.70 mmol) and 4 Å molecular sieves (1.00 g) in CH2Cl2 (15 mL) was treated with PCC (660 mg, 3.06 mmol) at room temperature. After stirring for 1.5 h at room temperature, the resulting suspension was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. The residue was purified by flash silica gel column chromatography (4 : 1 hexanes/EtOAc) to afford 27 (563 mg, 96%) as an amorphous yellow solid: [α]D25 +66.1 (c=1.0, CHCl3); IR (neat) cm−1: 3020, 1773, 1639, 1215; 1H-NMR (400 MHz, CDCl3) δ: 3.50 (d, 1H, J=10.4 Hz), 3.27 (d, 1H, J=10.4 Hz), 2.82 (dd, 1H, J=18.0, 10.0 Hz), 2.61 (dd, 1H, J=18.8, 7.6 Hz), 2.50–2.41 (m, 3H), 1.77 (dd, 1H, J=12.2, 7.6 Hz), 1.64–1.01 (m, 6H), 0.91 (s, 3H), 0.88 (s, 3H), 0.77 (s, 3H); 13C-NMR (400 MHz, CDCl3) δ: 204.3, 174.0, 86.1, 54.6, 48.0, 41.3, 39.3, 36.2, 36.1, 33.1, 32.2, 30.0, 21.1, 17.8, 13.6, 10.3; HR-MS (ESI) [M+Na]+ Calcd for C16H23NaIO3 413.0590. Found 413.0600.
(2R,3aR,4S,5aS,9aS,9bR)-3a-(Iodomethyl)-6,6,9a-trimethyldodecahydronaphtho[2,1-b]furan-2,4-diol (22)A solution of 27 (50.6 mg, 0.130 mmol) in CH2Cl2 (1.3 mL) was treated with DIBAL (1.02 M in n-hexane, 458 µL, 0.467 mmol) at −78°C. After stirring for 0.5 h at −78°C, the reaction mixture was quenched with MeOH. The organic layer was washed sequentially with 1 M HCl and H2O, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (4 : 1 hexanes/EtOAc) to afford 22 (32.1 mg, 63%) as a colorless amorphous solid: [α]D25 –27.0 (c=1.0, CHCl3); IR (neat) cm−1: 3486, 2932, 1423, 1215, 1061; 1H-NMR (400 MHz, CDCl3) δ: 5.50 (t, 1H, J=6.0 Hz), 4.06 (dd, 1H, J=11.6, 6.4 Hz), 3.70 (d, 1H, J=10.4 Hz), 3.62 (d, 1H, J=10.4 Hz), 2.28 (d, 1H, J=7.6 Hz), 2.22 (dd, 1H, J=14.2, 6.0 Hz), 1.99 (ddd, 1H, J=14.2, 7.6, 6.0 Hz), 1.81 (ddd, 1H, J=11.6, 6.4, 0.8 Hz), 1.57–0.80 (m, 8H), 0.91 (s, 3H), 0.86 (s, 3H), 0.84 (s, 3H); 13C-NMR (400 MHz, CDCl3) δ: 99.1, 85.9, 71.4, 56.5, 49.5, 41.4, 40.8, 35.8, 35.3, 33.6, 32.8, 27.8, 22.3, 18.1, 15.7, 12.9; HR-MS (ESI) [M+Na]+ Calcd for C16H27NaIO3 417.0903. Found 417.0904.
(1R,2S,3S,4aS,8aS)-1-Allyl-5,5,8a-trimethyloctahydro-1H-spiro[naphthalene-2,2′-oxiran]-3-ol (29)To a suspension of Ph3PMeBr (214 mg, 0.600 mmol) in THF (0.6 mL) was added dropwise n-BuLi (1.63 M in THF, 367 µL, 0.600 mmol) at −78°C. After stirring for 1 h at 0°C, a solution of 22 (33.7 mg, 0.0855 mmol) in THF (0.5 mL) was added. After stirring for an additional 1 h at 0°C, the reaction mixture was quenched with a saturated aq NH4Cl solution and extracted with EtOAc. The combined organic extracts were washed with H2O, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (12 : 1 hexanes/EtOAc) to afford 29 (20.0 mg, 89%) as a colorless amorphous solid: [α]D25 +36.3 (c=1.0, CHCl3); IR (neat) cm−1: 3476, 3078, 2958, 2928, 1521, 1398, 1215, 1082; 1H-NMR (400 MHz, CDCl3) δ: 5.70 (m, 1H), 4.97–4.92 (m, 2H), 3.66 (dt, 1H, J=11.6, 5.2 Hz), 2.87 (d, 1H, J=4.8 Hz), 2.62 (d, 1H, J=4.8 Hz), 2.05–2.00 (m, 2H), 1.92 (br d, 1H, J=11.6 Hz), 1.77–1.63 (m, 2H), 1.62–0.90 (m, 8H), 0.90 (s, 3H), 0.84 (s, 3H), 0.83 (s, 3H); 13C-NMR (400 MHz, CDCl3) δ: 140.1, 114.7, 69.3, 60.0, 52.4, 50.4, 44.9, 41.9, 39.4, 39.0, 33.4, 33.3, 30.6, 25.5, 21.6, 18.5, 14.6; HR-MS (ESI) [M+Na]+ Calcd for C17H28NaO2 287.1987. Found 287.1973.
(1R,2S,3S,4aS,8aS)-5,5,8a-Trimethyl-1-((Z)-3-methylpenta-2,4-dien-1-yl)octahydro-1H-spiro[naphthalene-2,2′-oxiran]-3-ol (21)A solution of 29 (29.6 mg, 0.112 mmol) in CH2Cl2 (2.2 mL) was treated with methacrolein (173 µL, 1.68 mmol) and a catalytic amount of Grubbs second-generation catalyst (9.5 mg, 0.0112 mmol) at room temperature. After stirring for 4.5 h at reflux, the reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was semi-purified by flash silica gel column chromatography (3 : 1 hexanes/EtOAc) to afford crude 28. To a suspension of Ph3PMeBr (140 mg, 0.392 mmol) in THF (0.6 mL) was added dropwise n-BuLi (1.60 M in THF, 245 µL, 0.392 mmol) at −78°C. After stirring for 1 h at 0°C, a solution of crude 28 in THF (0.5 mL) was added. After stirring for an additional 1 h at 0°C, the reaction mixture was quenched with a saturated aq NH4Cl solution and extracted with EtOAc. The combined organic extracts were washed with H2O, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (12 : 1 hexanes/EtOAc) to afford 21 (20.0 mg, 2 steps, 74%) as a colorless amorphous solid: [α]D25 +38.9 (c=1.0, CHCl3); IR (neat) cm−1: 3553, 3020, 2931, 2870, 1638, 1213, 1077; 1H-NMR (400 MHz, CDCl3) δ: 6.29 (dd, 1H, 17.6, 10.8 Hz), 5.28 (t, 1H, J=6.2 Hz), 5.06 (d, 1H, J=17.6 Hz), 4.91 (d, 1H, J=10.8 Hz), 3.67 (dt, 1H, J=11.6, 5.2 Hz), 2.87 (d, 1H, J=4.4 Hz), 2.47 (d, 1H, J=4.4 Hz), 2.06 (m, 2H), 1.85 (d, 1H, J=1.2 Hz), 1.85–1.77 (m, 2H), 1.71 (s, 3H), 1.64–0.82 (m, 8H), 0.91 (s, 3H), 0.88 (s, 3H), 0.85 (s, 3H); 13C-NMR (400 MHz, CDCl3) δ: 141.1, 134.6, 133.8, 110.7, 69.3, 60.0, 52.3, 52.2, 44.8, 41.8, 39.3, 39.2, 33.4, 33.3, 30.5, 21.6, 20.6, 18.5, 14.7, 12.0; HR-MS (ESI) [M+Na]+ Calcd for C20H32NaO2 327.2300. Found 327.2298.
Revised Structure of Cyslabdan (2)A solution of 21 (269 mg, 0.870 mmol) in MeOH–H2O (1 : 1, 35 mL) was treated with N-acetyl-L-cysteine (710 mg, 4.35 mmol) and Na2CO3 (1.38 mg, 13.0 mmol) at room temperature. After stirring for 15 h at 40°C, the reaction mixture was quenched with 2 M HCl and extracted with EtOAc. The organic layer was washed with H2O, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash silica gel column chromatography (10 : 1 CH2Cl2/MeOH) to afford 2 (353 mg, 87%) as a colorless amorphous solid: [α]D26 +32.4 (c=0.1, MeOH), (lit.4) [α]D25 +26.8 (c=0.1, MeOH); IR (neat) cm−1: 3490, 2949, 2834, 1654, 1411, 1201, 1019; 1H-NMR (400 MHz, CD3OD) δ: 6.34 (dd, 1H, J=17.6, 10.8 Hz), 5.52 (t, 1H, J=6.8 Hz), 5.04 (d, 1H, J=17.6 Hz), 4.86 (d, 1H, J=10.8 Hz), 4.47 (dd, 1H, J=7.6, 4.8 Hz), 3.76 (dd, 1H, J=11.2, 4.8 Hz), 3.03 (dd, 1H, J=13.2, 4.8 Hz), 2.82 (dd, 1H, J=13.2, 7.6 Hz), 2.78 (d, 1H, J=12.4 Hz), 2.62 (d, 1H, J=12.4 Hz), 2.42 (dt, 1H, J=17.6, 6.8 Hz), 2.11 (dd, 1H, J=17.6, 3.6 Hz), 1.98 (s, 3H), 1.75 (br s, 3H), 1.73–0.84 (m, 10H), 1.00 (s, 3H), 0.92 (s, 3H), 0.87 (s, 3H); 13C-NMR (400 MHz, CD3OD) δ: 176.0, 173.1, 143.0, 137.5, 133.4, 110.2, 78.7, 72.8, 55.2, 54.7, 53.8, 43.0, 41.0, 39.8, 39.1, 37.3, 34.1, 34.0, 27.9, 24.3, 22.6, 22.3, 19.3, 15.9, 12.0; HR-MS (ESI) [M+Na]+ Calcd for C25H41NNaO5 490.2603. Found 490.2600.