2. Reversing Reactivities of Carbonyl Functions in Reduction and Alkylation
2.1. Selective Transformations of Less Reactive Carbonyl Groups in the Presence of AldehydesWe developed a new chemoselective method for the deprotection of acetals in the presence of ketals using a combination of 2,4,6-collidine and triethylsilyl trifluoromethanesulfonate (TESOTf)23–25) (Chart 1, Eq. 1). This is the only method reported to date for the deprotection of acetals in the presence of ketals; in conventional methods, ketal deprotection is faster than acetal deprotection.26) The key aspect of the reaction is the selective formation of collidinium salt intermediates from acetals by distinguishing their steric environments. In an earlier study, we achieved nucleophilic substitution reactions of the salt intermediates with various nucleophiles25,27) including Gilman reagents, in which the C–C bond forming activities of acetals and ketals were reversed (Chart 1, Eq. 2) compared with those in conventional methods. However, alkyllithium reagents did not react well with the salts.27)
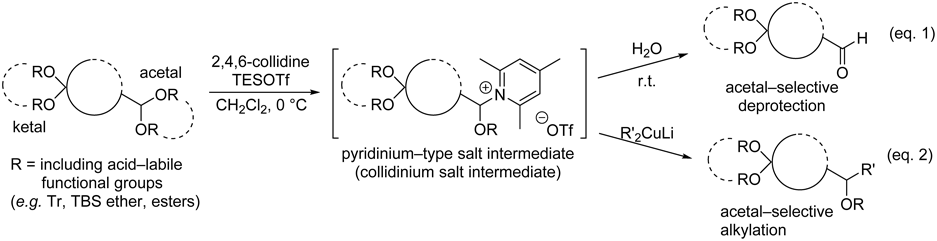
Chart 1. Chemoselective Transformation of Acetals in the Presence of Ketals
We then assumed that chemoselective transformations of ketones and aldehydes could proceed as shown in Chart 2, i.e., the selective formation of a salt of an aldehyde in a keto-aldehyde and addition of alkyllithium reagents, as a hard nucleophile, to the aldehyde–salt intermediates would result in reaction with the ketone, leaving the pyridinium salt intact. On the other hand, the addition of Gilman reagents, as a soft nucleophile, to the the aldehyde–salt intermediates would result in nucleophilic substitution with the pyridinium salt selectively, leaving the ketone unit intact (Chart 2).

Chart 2. Strategy for the Selective Transformation via Pyridinium-Type Salt Intermediates
However, the reaction of keto-aldehyde 1 with TESOTf and 2,4,6-collidine afforded a complex mixture perhaps because of the basicity of 2,4,6-collidine. One of the features of our salt method is the use of various types of silyl trifluoromethanesulfonates (triflate) and pyridine bases. When quinoline was used instead of 2,4,6-collidine and tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) was used instead of TESOTf, followed by addition of PhLi, selective transformation of the ketone unit was achieved in the presence of the aldehyde unit in keto-aldehyde 1. However, the yield of the desired product was moderate. In this case, a silyl enol ether was also generated by enolization of the ketone unit as a consequence of the basicity of quinoline28) (Chart 3).

Chart 3. Reaction of Keto-Aldehyde 1
In a continuation of these studies, we used phosphines instead of pyridines for the salt-forming process; this makes the reaction conditions weakly acidic and controls ketone enolization.29,30) We had already observed that the reactivities of phosphonium salt intermediates, which were generated from O,O-acetals and various phosphines, are similar to those of pyridinium salt intermediates.31) We also found that the phosphonium salt reactivity strongly depends on the type of phosphine.32) The phosphonium salts of tris(o-tolyl)phosphine (P(o-Tol)3) (Tol=tolyl) actively participate in Grignard reactions to afford the desired products in high yields in short reaction times; the phosphonium salts from triphenylphosphine (PPh3) are less reactive and the yields of the desired products are low (Chart 4). We then developed a strategy for reversing the reactivities of keto-aldehydes using PPh3 as the phosphine, as shown in Chart 5.33,34)

Chart 4. Grignard Reaction of Phosphonium Salts of PPh3 and P(o–Tol)3
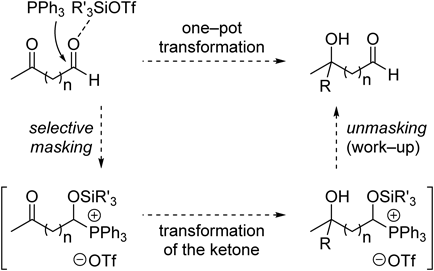
Chart 5. Strategy for Reversing Reactivities of Carbonyl Functions Using Phosphonium Salt Intermediates
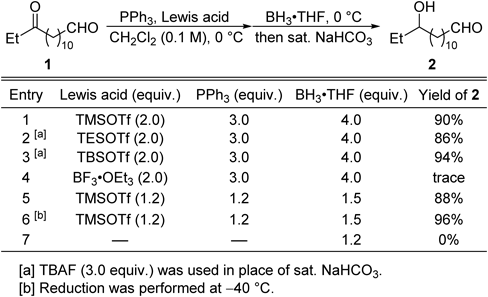
We investigated various Lewis acid/PPh3 combinations and BH3·tetrahydrofuran (THF) reduction (Table 1). The selective transformation of a ketone to an alcohol in the presence of an aldehyde was achieved using a PPh3–silyl triflate combination (Table 1, entries 1–3). When trimethylsilyl trifluoromethanesulfonate (TMSOTf) was used, the aldehyde was recovered from the phosphonium salt by hydrolysis with a weak base (aq. NaHCO3; Table 1, entry 1). In contrast, when other silyltriflates such as TESOTf and TBSOTf were used, treatment with tetrabutylammonium fluoride (TBAF, 3.0 equiv.) was necessary to recover the aldehyde (Table 1, entries 2, 3). The use of BF3·Et2O was not effective in this reaction (Table 1, entry 4). Reducing the amounts of PPh3 and TMSOTf did significantly lower the yield, but reduction at −40°C gave the best results (Table 1, entries 5, 6). Nonselective reduction occurred in the absence of the silyltriflate and PPh3, and a diol and aldehyde-reduced keto alcohol were obtained in 20 and 74% yields, respectively (Table 1, entry 7).
Table 1. Optimization of Reaction Conditions
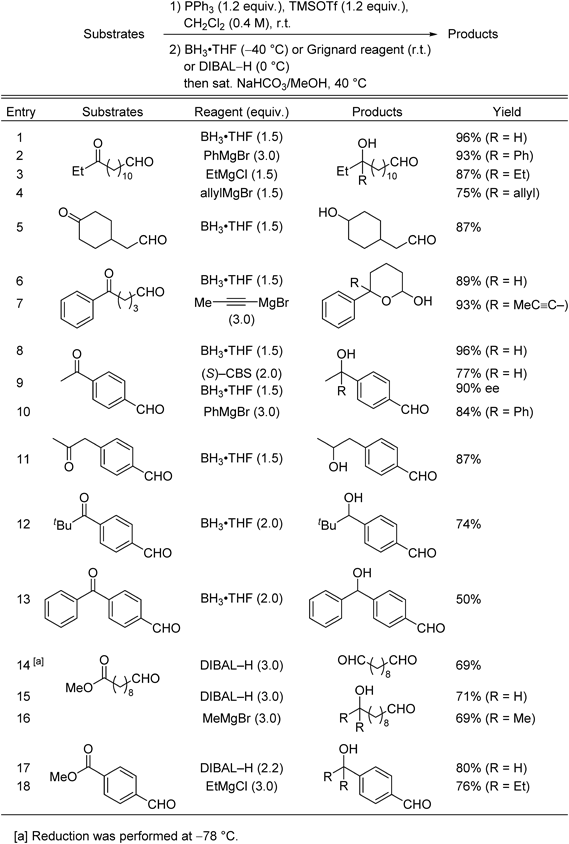
This method was successfully applied to substrates containing two different carbonyl groups, i.e., ketones or esters and aldehydes, in the same molecule.33) The in situ protection method, which proceeds via O,P-acetal phosphonium salt intermediates,34) can be used in alkylation with Grignard reagents and reduction with BH3·THF or diisobutylaluminum hydride (DIBAL–H) (Table 2). The results show that the in situ protection method enabled selective ketone reduction and alkylation of keto-aldehyde substrates (Table 2, entries 1–13). The high-yield transformations of the 5-oxoaldehyde derivative (Table 2, entries 6, 7) show that this method can be used for the efficient construction of synthetically useful lactols. Aromatic aldehydes also participated in the selective in situ protection using phosphonium salts (Table 2, entries 8–13). Use of the Corey–Bakshi–Shibata (CBS) reagent35) enabled asymmetric reduction of a ketone in the presence of an aldehyde, with good selectivity (Table 2, entry 9). It is worth noting that the ketone group in a readily enolizable ketone was selectively reduced in high yield, showing that the protection/deprotection conditions used in this method are very mild (Table 2, entry 11). A substrate with a bulky ketone group was also selectively converted to the corresponding alcohol when 2 equiv. of the reductant BH3·THF were used (Table 2, entry 12). A benzophenone ketone was reduced to generate the desired product in only moderate yield because over-reduction of the product produced 4-benzylbenzaldehyde in 17% yield (Table 2, entry 13). Ester groups in substrates that also contain aldehyde moieties can also be selectively transformed to primary alcohols by DIBAL–H to tertiary alcohols by Grignard addition using the in situ protection method (Table 2, entries 14–18). The reaction of DIBAL–H with an ester-aldehyde at −78°C under in situ protection conditions, produced the semi-reduced dialdehyde in good yield (Table 2, entry 14).
Table 2. Selective Transformation of Less Reactive Carbonyl Functions in the Presence of Aldehyde
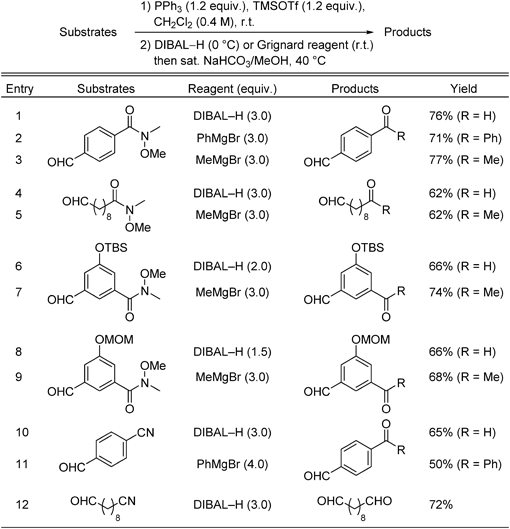
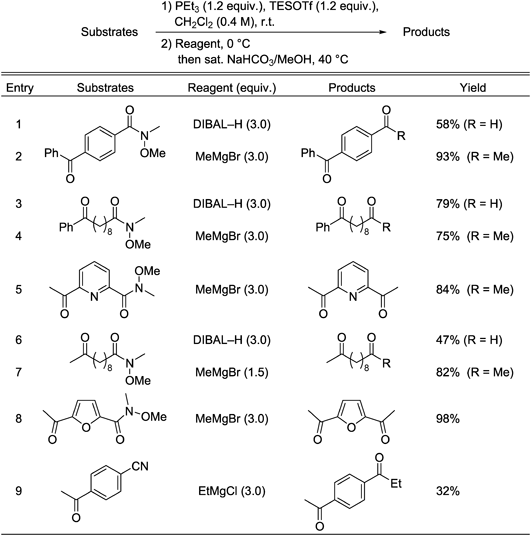
The results listed in Table 3 show that the in situ protection method enables selective transformations of amide and nitrile groups in substrates that also contain aldehyde moieties. Reduction and Grignard addition reactions of Weinreb amides occurred smoothly to yield the respective dialdehydes and keto-aldehydes (Table 3, entries 1–5). tert-Butyldimethylsilyl (TBS) and methoxymethyl (MOM) groups tolerate the in situ protection/deprotection conditions (Table 3, entries 6–9). The nitrile groups in nitrile-aldehydes underwent selective reduction with DIBAL–H and Grignard addition to afford the corresponding dialdehyde and keto-aldehyde products (Table 3, entries 10–12).
Table 3. Selective Transformations of Weinreb Amides or Nitriles in the Presence of Aldehydes
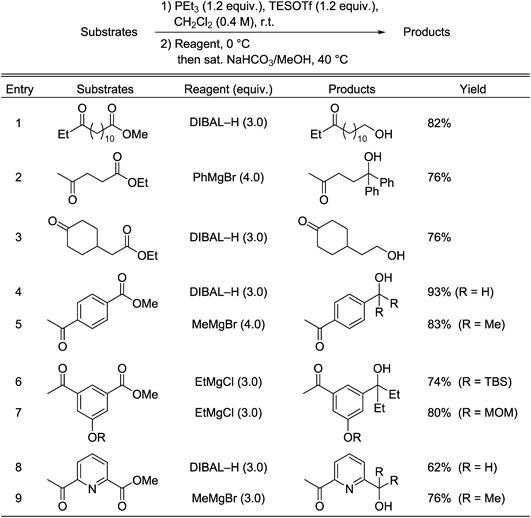
A combination of PPh3 and TMSOTf only converts aldehydes, not ketones or esters, to the corresponding O,P-acetal phosphonium salts, even when excess amounts of the reagents are used (see Table 1, entry 1). Ketones do not react with PPh3–TMSOTf because of the steric bulkiness and low nucleophilicity of PPh3. We therefore used PEt3, which is highly reactive and commercially available, instead of PPh3. If PEt3 is used instead of PPh3, the above method can be used in the selective in situ protection of ketones. The salt intermediate was successfully formed using PEt3 (Chart 6). Table 4 shows the results for PEt3-based in situ ketone protection in ester-selective reactions of keto-esters. Various keto-esters, including a substrate containing a cyclohexanone moiety, reacted with DIBAL–H and Grignard reagents to furnish the corresponding primary and tertiary alcohols, respectively; the reaction involves initial formation of ketone-protected phosphonium ketals. It is worth noting that keto-ester substrates containing TBS and MOM protecting groups and a pyridine heterocyclic ring underwent selective reactions at their ester centers (Table 4, entries 6–9).
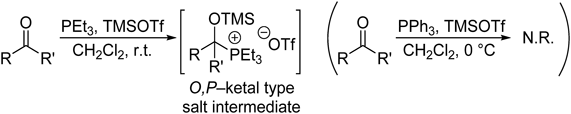
Chart 6. O,P-Ketal Phosphonium Salt Formation from Ketones and PEt3
Table 4. Selective Transformation of Esters in the Presence of Ketones
This method was used in the selective transformation of substances containing ketone moieties along with either Weinreb amide or nitrile groups (Table 5). The results show that Weinreb amides generally participate in efficient reduction and alkylation reactions of these substrates, but the reductions gave low yields (Table 5, entries 1, 6). The nitrile used in the reaction shown in entry 9 was not effective in this reaction and only 32% of the desired product was obtained (Table 5, entry 9).
Table 5. Selective Transformation of Weinreb Amide or Nitrile in the Presence of Aldehyde
(+)-Centrolobine, which is isolated from the heartwood of Centrolobium robustum, has anti-inflammatory, antibacterial, and anti-leishmanial activities.36–38) Asymmetric syntheses of centrolobine have been reported by several groups, but they involve multiple steps and/or give low yields.39–55) Our synthetic route is summarized in Chart 7. This enantioselective synthesis was accomplished in five steps from commercially available cyclopentene 3; the overall yield was 75%, which is the highest total yield reported for the asymmetric synthesis of (+)-centrolobine. Our in situ protection method was used in the conversion of keto-aldehyde 4 to lactol 5. Asymmetric ketone reduction was achieved using CBS reagent.35)

Chart 7. Asymmetric Total Synthesis of (+)-Centrolobine Using in Situ Protection Method
To the best of our knowledge, this is the first and only example of asymmetric transformation of a ketone in the presence of an aldehyde.
3. Control of Carbonyl Function Reactivity in Reduction and Alkylation56,57)
3.1. Selective Transformation of Similar Reactive Carbonyl Groups in the Presence of α,β-Unsaturated Ketones (Enones)Ketones and α,β-unsaturated ketones (enones) display nearly identical reactivities, although the reactivities of ketones toward nucleophiles are usually slighter higher than those of enones. Three different methods have been reported for selective reduction of ketones in the presence of enones,58–60) and there is no general method for the selective reduction of carbonyl functions in the presence of enones, or for selective alkylation.
As described in Section 2.1., we found that a combination of PPh3 and TMSOTf, even when excess PPh3 is used, does not transform ketones to their corresponding phosphonium salts. However, other groups have reported that β-alkylation reactions of enones using PPh3 and TBSOTf occur via initial phosphoniosilylation.61–63) We therefore thought that a combination of PPh3 and a silyl triflate would react selectively with enones in the presence of ketones. We presumed that if the phosphonium silyl enol ethers could survive during transformation of the remaining ketones, an in situ protection method for discriminating between closely related functional groups could be established as shown in Chart 8.

Chart 8. Strategy for Control of Reactivity of Carbonyl Functions via Phosphonium Salt Intermediates
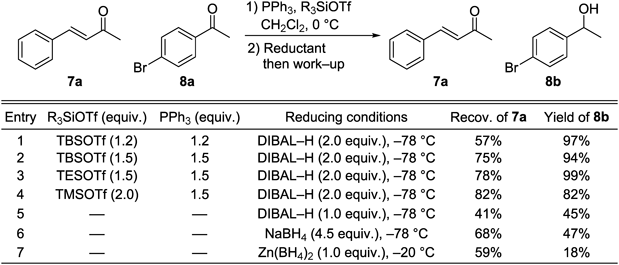
The optimum reaction conditions for discrimination between enones and ketones were identified using enone 7a and ketone 8a (Table 6). In the light of previous reports,61–63) we chose TBSOTf and PPh3 as the reagents for enone protection and DIBAL–H as the reducing agent (Table 6, entry 1). Treatment of a 1 : 1 mixture of 7a and 8a with TBSOTf and PPh3 (1.2 equiv. each) and then with DIBAL–H, followed by TBAF workup, gave the ketone-reduced alcohol 8b (97%) along with recovered enone 7a (57%). Increasing the amounts of PPh3 and TBSOTf to 1.5 equiv. afforded a higher yield of recovered 7a (75%) (Table 6, entry 2). The use of TESOTf instead of TBSOTf resulted in a slightly higher 7a recovery (78%) (Table 6, entry 3). A higher yield of recovered 7a (82%) was obtained with TMSOTf and weakly basic solvolysis conditions (K2CO3–MeOH) instead of TBAF workup (Table 6, entry 4). Nonselective reduction occurred in the absence of pretreatment with PPh3 and a silyl triflate (Table 6, entry 5). Two previously reported methods62,63) were not effective with these aromatic substrates (Table 6, entries 6, 7).
Table 6. Optimization of Reaction Conditions
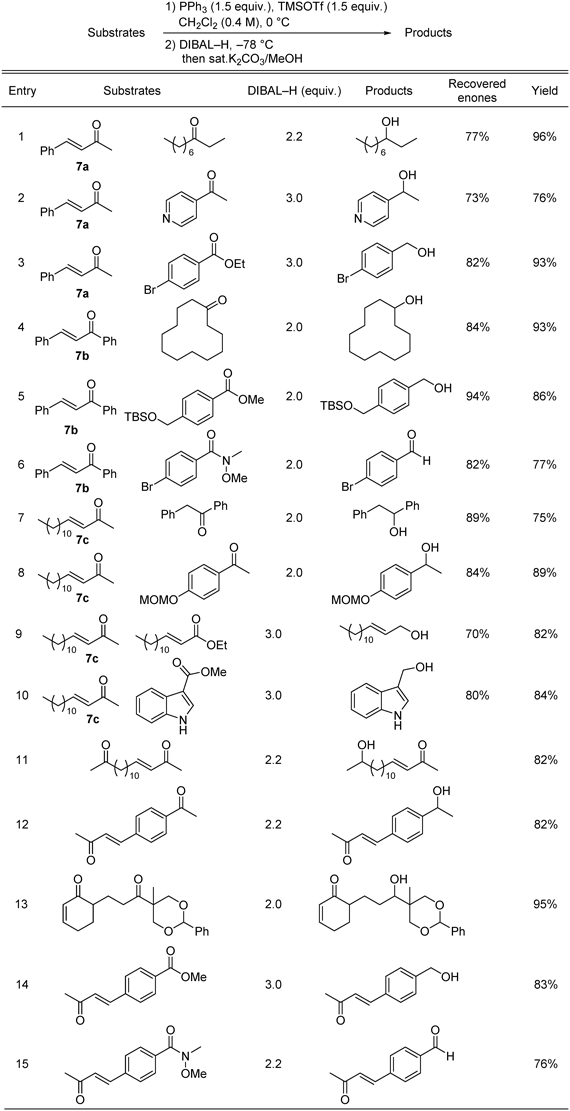
We explored the generality of this method (Table 7). The results show that pretreatment with PPh3 and TMSOTf can be used to protect aromatic and aliphatic enones in selective reduction of various ketones (Table 7, entries 1, 4, 8) and easily enolizable ketone (Table 7, entry 7). The selective reduction of esters (Table 7, entries 3, 5), including an α,β-unsaturated ester (Table 7, entry 9) and Weinreb amide (Table 7, entry 6), proceeded smoothly to produce the corresponding alcohols and aldehyde in high yields (Table 7, entries 1, 3–9). A pyridyl ketone and unprotected indolyl ester were selectively reduced (Table 7, entries 2, 10). The acid-labile TBS and MOM groups tolerated the reaction conditions (Table 7, entries 5, 8). The reaction was used in the selective reduction of substrates containing both enone and other carbonyl moieties in the same molecule (Table 7, entries 11–15). The ketone groups in aliphatic and aromatic keto-enones were reduced selectively to afford the corresponding alcohols in high yields (Table 7, entries 11, 12). Even a highly hindered ketone moiety that also contained an acid-labile benzylidene acetal group was reduced, to give an excellent product yield, leaving the enone and acetal groups intact (Table 7, entry 13). Ester and Weinreb amide moieties were also reduced in the presence of enone groups (Table 7, entries 14, 15).
Table 7. Selective Reduction of Various Carbonyl Functions in the Presence of Enones
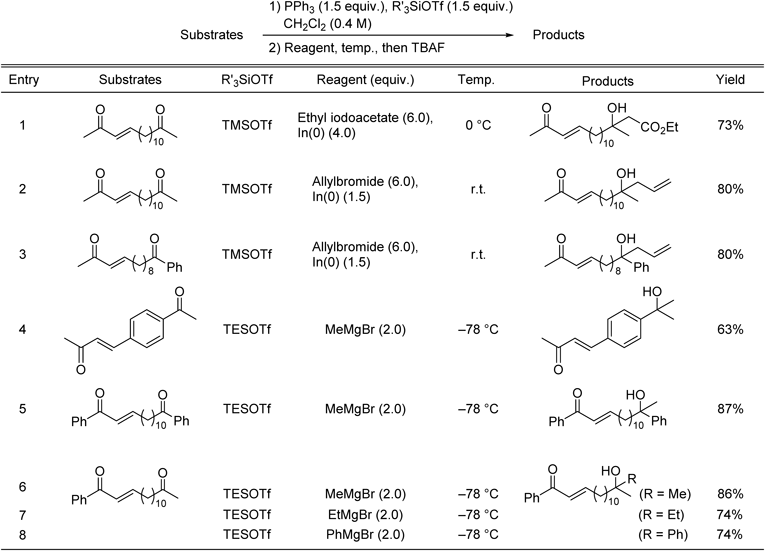
It is worth noting that the in situ protection method can also be applied to ketone-selective alkylation of keto-enones (Table 8). Reformatsky-type64) and Grignard reactions afforded the ketone-selective alkylated products, respectively (Table 8, entries 1, 4–8). In(0)-mediated Barbier-type allylation65) also selectively react with ketones, affording the corresponding alcohols in high yields (Table 8, entries 2, 3). These are the first reported methodsfor ketone-selective alkylation of keto-enones.
Table 8. Selective Alkylations of Various Carbonyl Functions in the Presence of Enones
Decytospolides A and B were isolated from Cytospora sp., an endophytic fungus from Ilex canariensis.66) Decytospolide B shows in vitro cytotoxic activity toward tumor cell lines A549 and QGY. There are two reports of the total synthesis of these natural products; the synthetic routes involve multiple steps.67,68) Our synthetic route to the target natural products is summarized in Chart 9. Our in situ protection method achieved the conversion of optically active keto-enone 11, obtained from the commercially available cyclopentanone 9, by asymmetric reduction with the CBS reagent,35) a subsequent two-step sequence gave oxacyclic compound 12. The side chain was isomerized using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). The enantioselective syntheses of decytospolides A and B from 9 were accomplished in six and seven steps, respectively, in overall yields of 47 and 46%, respectively.

Chart 9. Asymmetric Total Synthesis of Decytospolides A and B