Experimental
Chemicals, Reagents and InstrumentsChemicals were obtained from Sigma-Aldrich, U.S.A. and were used without further purification. Melting points were determined on a Stuart SMP10 capillary melting point apparatus and are uncorrected. TLC was used to monitor the progress and/or completion of the reactions using a pre-coated sheet (Eastman Kodak Co., Silca 60 F254) and was visualized with UV light at 254 nm. Elemental analysis was performed by Micro Analytical Center, Faculty of Science, Cairo University, Giza, Egypt and were within ±0.4% of the calculated values. 1H-NMR was obtained using a Bruker Advance-II 400 Spectrometer on 400 MHz using tetramethylsilane (TMS) as internal standard.
Chemistry. General Procedure for the Synthesis of Symmetrical Esters of Dialkyl 4-(Nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (3a–l)To a 50-mL round-bottomed flask, ammonium acetate (0.162 g, 2.10 mmol) was added to a stirring solution of 2-nitrobenzaldehyde or 3-nitrobenzaldehyde (0.254 g, 1.65 mmol) and the corresponding alkyl acetoacetate (3.35 mmol) in methanol or 2-propanol (10 mL). The reaction mixture was protected from light and heated under reflux for 12–24 h. After cooling, the precipitate was filtered and purified by crystallization from methanol or 2-propanol to afford the corresponding product.
Dimethyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3a)Recrystallization from methanol afforded (0.480 g, 79%): melting point (mp): 186–188°C.22)
Diethyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3b)Recrystallization from methanol afforded 0.434 g (66%): mp: 128–130°C.22)
Diisopropyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3c)Recrystallization from methanol afforded (0.358 g, 54%): Rf=0.65 (CHCl3–MeOH, 95 : 5); mp: 132–134°C; 1H-NMR (400 MHz, CDCl3) δ: 0.67 (d, J=5.9, 6.4 Hz, 6H, CH(CH3)2), 1.02 (d, J=6.4 Hz, 6H, CH(CH3)2), 2.41 (s, 6H, C2-CH3, C6-CH3), 4.72–4.78 (m, J=6.0 Hz, 2H, 2 of CH(CH3)2), 5.31 (s, 1H, C4-H), 5.51 (s, 1H, NH), 7.61 (t, J=7.5 Hz, 1H, aromatic C(5′)H), 7.77(t, J=8.4 Hz, 1H, aromatic C(4′)H), 7.86 (d, J=7.5 Hz, 1H, aromatic C(6′)H), 8.08 (d, J=8.4 Hz, 1H, aromatic C(3′)H); Anal. (%): (C21H26N2O6), Calcd (Found): C 62.67 (62.68), H 6.51 (6.45), N 6.96 (7.10).
Diisobutyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3d)Recrystallization from 2-propanol afforded (0.412 g, 58%): Rf=0.69 (CHCl3–MeOH, 95 : 5); mp: 135–137°C; 1H-NMR (400 MHz, CDCl3) δ: 0.79 (d, J=6.8 Hz, 6H, CH(CH3)2), 0.83 (d, J=6.8 Hz, 6H, CH(CH3)2), 1.80–1.86 (m, J=6.4 Hz, 6.8 Hz, 2H, 2 of CH(CH3)2), 2.31 (s, 6H, C2-CH3, C6-CH3), 3.76 (d, J=6.4 Hz, 4H, 2 of COOCH2), 5.08 (s, 1H, C4-H), 5.66 (s, 1H, NH), 7.19 (t, J=8.0 Hz, 1H, aromatic C(5′)H), 7.31 (t, J=7.6, 8.0 Hz, 1H, aromatic C(4′)H), 7.58 (d, J=8.0 Hz, 1H, aromatic C(6′)H), 7.93 (d, J=7.6 Hz, 1H, aromatic C(3′)H); Anal. (%): (C23H30N2O6), Calcd (Found): C 64.17 (64.07), H 7.02 (6.74), N 6.51 (6.55).
Bis(2-methoxyethyl) 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3e)Recrystallization from methanol afforded (0.588 g, 82%): Rf=0.38 (CHCl3–MeOH, 95 : 5); mp: 124–126°C; 1H-NMR (400 MHz, CDCl3) δ: 2.36 (s, 6H, C2-CH3, C6-CH3), 3.36 (s, 6H, 2 of OCH3), 3.54–3.57 (m, J=4.0, 4.8 Hz, 4H, 2 of COOCH2CH2O), 4.13–4.24 (m, J=4.0, 4.8 Hz, 4H, 2 of COOCH2), 5.14 (s,1H, C4-H), 6.07 (s, 1H, NH), 7.28 (t, J=8.0 Hz, 1H, aromatic C(5′)H), 7.38 (t, J=7.6, 8.0 Hz, 1H, aromatic C(4′)H), 7.70 (d, J=8.0 Hz, 1H, aromatic C(6′)H), 8.02 (d, J=7.6 Hz, 1H, aromatic C(3′)H); Anal. (%): (C21H26N2O8), Calcd (Found): C 58.06 (57.80), H 6.03 (6.11), N 6.45 (6.64).
Dibenzyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3f)Recrystallization from 2-propanol afforded (0.525 g, 65%): Rf=0.89 (CHCl3–MeOH, 95 : 5); mp: 102–104°C; 1H-NMR (400 MHz, CDCl3) δ: 1.65 (s, 1H, NH), 2.39 (s, 6H, C2-CH3, C6-CH3), 5.04 (d, J=5.50 Hz, 2H, COOCH2), 5.26 (d, J=5.50 Hz, 2H, COOCH2), 5.87 (s, 1H, C4-H), 7.20–7.26 (m, 11H, aromatic OCH2C6H5), 7.31 (t, J=8.4 Hz, 1H, aromatic C(5′)H), 7.38 (t, J=7.6, 8.4 Hz, 1H, aromatic C(4′)H), 7.48 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 8.97 (d, J=8.4 Hz, 1H, aromatic C(3′)H); Anal. (%): (C29H26N2O6), Calcd (Found): C 69.87 (69.62), H 5.26(4.98), N 5.62 (6.06).
Dimethyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3g)Recrystallization from methanol afforded (0.364 g, 60%), mp: 188–190°C.22)
Diethyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3h)Recrystallization from methanol afforded 0.355 g (54%); mp: 132–134°C.22)
Diisopropyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3i)Recrystallization from methanol afforded (0.465 g, 70%): Rf=0.79 (CHCl3–MeOH, 95 : 5); mp: 116–118°C; 1H-NMR (400 MHz, CDCl3) δ: 1.11 (d, J=6.0 Hz, 6H, CH(CH3)2), 1.28 (d, J=6.0 Hz, 6H, CH(CH3)2), 2.36 (s, 6H, C2-CH3, C6-CH3), 4.92–5.00 (m, J=6.0, 6.4 Hz, 2H, 2 of CH(CH3)2), 5.08 (s, 1H, C4-H), 5.87 (s, 1H, NH), 7.38 (t, J=7.6, 8.0 Hz, 1H, aromatic C(5′)H), 7.65 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 8.02 (d, J=8.0 Hz, 1H, aromatic C(4′)H), 8.15 (s, 1H, aromatic C(2′)H)); Anal. (%): (C21H26N2O6), Calcd (Found): C 62.67 (62.68), H 6.51 (6.45), N 6.96 (7.10).
Diisobutyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3j)Recrystallization from 2-propanol afforded (0.376 g, 53%): Rf=0.82 (CHCl3–MeOH, 95 : 5); mp: 95–97°C; 1H-NMR (400 MHz, CDCl3) δ: 0.88 (d, J=6.8 Hz, 6H, CH(CH3)2), 0.92 (d, J=6.8 Hz, 6H, CH(CH3)2), 1.87–1.97 (m, J=6.4, 6.8 Hz, 2H, 2 of CH(CH3)2), 2.39 (s, 6H, C2-CH3, C6-CH3), 3.85 (d, J=6.4 Hz, 4H, 2 of COOCH2), 5.16 (s, 1H, C4-H), 5.99 (s, 1H, NH), 7.39 (t, J=7.6, 8.0 Hz, 1H, aromatic C(5′)H), 7.68 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 8.02 (d, J=8.0 Hz, 1H, aromatic C(4′)H), 8.15 (s, 1H, aromatic C(2′)H); Anal. (%): (C23H30N2O6), Calcd (Found): C 64.17 (64.07), H 7.02 (6.74), N 6.51 (6.55).
Bis(2-methoxyethyl) 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3k)Recrystallization from methanol afforded (0.538 g, 75%): Rf=0.45 (CHCl3–MeOH, 95 : 5); mp: 118–120°C; 1H-NMR (400 MHz, CDCl3) δ: 2.35 (s, 6H, C2-CH3, C6-CH3), 3.34 (s, 6H, 2 of OCH3), 3.51–3.59 (m, J=4.00, 6.80 Hz, 4H, 2 of COOCH2CH2O), 4.12–4.22 (m, J=4.00, 7.4 Hz, 4H, 2 of COOCH2), 5.13 (s, 1H, C4-H), 6.23 (s, 1H, NH), 7.35–7.39 (t, J=7.6, 8.00. Hz, 1H, aromatic C(5′)H), 7.69 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 8.00 (d, J=8.0 Hz, 1H, aromatic C(4′)H), 8.13 (s, 1H, aromatic C(2′)H); Anal. (%): (C21H26N2O8), Calcd (Found): C 58.06 (57.78), H 6.03 (5.92), N 6.45 (6.63).
Dibenzyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3l)Recrystallization from 2-propanol afforded (0.420 g, 52%): Rf=0.91 (CHCl3–MeOH, 95 : 5); mp: 99–101°C; 1H-NMR (400 MHz, CDCl3) δ: 1.67 (s, 1H, NH), 2.38 (s, 6H, C2-CH3, C6-CH3), 5.04 (d, J=12.4 Hz, 2H, COOCH2), 5.12 (d, J=12.4 Hz, 2H, COOCH2), 5.88 (s, 1H, C4-H), 7.19–7.24 (m, 11H, aromatic OCH2C6H5), 7.31 (t, J=7.6, 8.0 Hz, 1H, aromatic C(5′)H), 7.48 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 7.97 (d, J=8.0 Hz, 1H, aromatic C(4′)H), 8.03 (s, 1H, aromatic C(2′)H); Anal. (%): (C29H26N2O6), Calcd (Found): C 69.87 (69.71), H 5.26 (5.11), N 5.62 (5.98).
General Procedure for the Synthesis of Asymmetrical Esters of Dialkyl 4-(Nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (6a–j)To a 50-mL round-bottomed flask were added a mixture of 2-nitrobenzaldehyde or 3-nitrobenzaldehyde (0.254 g, 1.65 mmol), alkyl acetoacetate (1.65 mmol) and alkyl 3-aminocrotonate (1.65 mmol) in methanol or 2-propanol (10 mL). The reaction mixture was protected from light and heated under reflux for 9–24 h. After cooling, the precipitate was filtered and purified by crystallization from methanol or 2-propanol to afford the corresponding product.
3-Ethyl 5-Methyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6a)Recrystallization from methanol afforded (0.216 g, 62%): Rf=0.76 (CHCl3–MeOH, 95 : 5); mp: 142–144°C; 1H-NMR (400 MHz, CDCl3) δ: 1.02 (t, J=7.4 Hz, 3H, CH2CH3), 1.58 (s, 6H, C2-CH3, C6-CH3), 2.51 (s, 3H, COOCH3), 3.95–4.03 (m, J=7.4 Hz, 2H, COOCH2), 5.37 (s, 1H, C4-H), 5.72 (s, 1H, NH), 7.46 (t, J=8.0 Hz, 1H, aromatic C(5′)H), 7.55 (t, J=7.6, 8.0 Hz, 1H, aromatic C(4′)H), 7.75 (d, J=8.0 Hz, 1H, aromatic C(6′)H), 7.95 (d, J=7.6 Hz, 1H, aromatic C(3′)H); Anal. (%): (C18H20N2O4), Calcd (Found): C 59.99 (59.74), H 5.59 (5.27), N 7.77 (8.00).
3-Ethyl 5-Isopropyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6b)Recrystallization from methanol afforded (0.455 g, 71%): Rf=0.73 (CHCl3–MeOH, 95 : 5); mp: 115–117°C; 1H-NMR (400 MHz, CDCl3) δ: 1.11 (d, J=6.0 Hz, 3H, CH(CH3)2), 1.21–1.27 (m, J=6.0, 7.2 Hz, 6H, CH(CH3)2 and CH2CH3), 2.36 (s, 6H, C2-CH3, C6-CH3), 4.05–4.14 (m, J=3.6, 7.2 Hz, 2H, COOCH2), 4.92–4.99 (m, J=6.0, 6.4 Hz, 1H, CH(CH3)2), 5.09 (s, 1H, C4-H), 6.10 (s, 1H, NH), 7.44 (t, J=8.0, 8.4 Hz, 1H, aromatic C(5′)H), 7.69(t, J=8.0 Hz, 1H, aromatic C(4′)H), 8.05 (d, J=8.4 Hz, 1H, aromatic C(6′)H), 8.18 (d, J=8.0 Hz, 1H, aromatic C(3′)H); Anal. (%): (C20H24N2O6), Calcd (Found): C 61.84 (62.00), H 6.23 (6.32), N 7.21 (7.27).
3-(2-Methoxyethyl) 5-Methyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6c)Recrystallization from methanol afforded (0.329 g, 51%): Rf=0.50 (CHCl3–MeOH, 95 : 5); mp: 110–112°C; 1H-NMR (400 MHz, CDCl3) δ: 2.30 (s, 6H, C2-CH3, C6-CH3), 3.30 (s, 6H, OCH3, COOCH3), 4.03–4.09 (m, J=3.6,4.0 Hz, 2H, COOCH2CH2O), 4.24–4.30 (m, 2H, J=4.0, 4.8 Hz COOCH2CH2O), 5.47 (s, 1H, C4-H), 5.90 (s, 1H, NH), 7.28 (t, J=6.0, 8.0 Hz, 1H, aromatic C(5′)H), 7.48 (t, J=7.2, 7.6 Hz, 1H, aromatic C(4′)H), 7.54 (d, J=7.2 Hz, 1H, aromatic C(6′)H), 7.75 (d, J=8.0 Hz, 1H, aromatic C(3′)H); Anal. (%): (C19H22N2O7), Calcd (Found): C 58.46 (58.32), H 5.68 (5.69), N 7.18 (7.11).
3-Ethyl 5-(2-Methoxyethyl) 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6d)Recrystallization from methanol afforded (0.440 g, 66%): Rf=0.53 (CHCl3–MeOH (95 : 5); mp: 105–107°C; 1H-NMR (400 MHz, CDCl3) δ: 1.17 (t, J=7.2 Hz, 3H, CH2CH3), 2.33 (s, 6H, C2-CH3, C6-CH3), 3.31 (s, 3H, OCH3), 3.50–3.62 (m, J=4.00,6.8 Hz, 2H, COOCH2CH2O), 3.96–4.17 (m, J=4.00, 6.8 Hz, 2H, COOCH2CH3), 4.24–4.29 (m, J=4.00, 4.8 Hz, 2H, COOCH2CH2O), 5.87 (s, 1H, C4-H), 5.89 (s, 1H, NH), 7.26 (t, J=7.6, 8.00 Hz, 1H, aromatic C(5′)H), 7.49 (t, J=7.2, 7.6 Hz, 1H, aromatic C(4′)H), 7.54 (d, J=8.00 Hz, 1H, aromatic C(6′)H), 7.74 (d, J=7.6 Hz, 1H, aromatic C(3′)H); Anal. (%): (C20H24N2O7), Calcd (Found): C 59.4 (59.13), H 5.98 (5.81), N 6.93 (6.92).
3-Benzyl 5-Methyl 2,6-Dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6e)Recrystallization from methanol afforded (0.481 g, 69%): Rf=0.70 (CHCl3–MeOH, 95 : 5); mp: 108–110°C; 1H-NMR (400 MHz, CDCl3) δ: 2.25 (s, 3H, COOCH3), 3.54 (s, 6H, C2-CH3, C6-CH3), 3.56 (s, 2H, COOCH2), 5.10 (s, 1H, C4-H), 6.11 (s, 1H, NH), 7.09–7.18 (m, 11H, aromatic OCH2C6H5), 7.22 (t, J=7.6 Hz, 1H, aromatic C(5′)H), 7.47 (t, J=7.6, 8.0 Hz, 1H, aromatic C(4′)H), 7.88 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 7.97 (d, J=8.0 Hz, 1H, aromatic C(3′)H); Anal. (%): (C23H22N2O6), Calcd (Found): C 65.39 (65.06), H 5.52 (5.26), N 6.63 (6.54).
3-Ethyl 5-Methyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6f)Recrystallization from methanol afforded (0.213 g, 59%): Rf=0.79 (CHCl3–CH3OH 95 : 5); mp: 162–164°C; 1H-NMR (400 MHz, CDCl3) δ: 1.10 (t, J=7.4 Hz, 3H, CH2CH3), 2.49 (s, 6H, C2-CH3, C6-CH3), 3.57 (s, 3H, COOCH3), 3.90–4.21 (m, J=7.4 Hz, 2H, COOCH2), 5.29(s, 1H, C4-H), 5.70 (s, 1H, NH), 7.39 (t, J=8.5 Hz, 1H, aromatic C(5′)H), 7.69 (d, J=7.5 Hz, 1H, aromatic C(6′)H), 8.00 (d, J=8.5 Hz, 1H, aromatic C(4′)H), 8.13 (s, 1H, aromatic C(2′)H); Anal. (%): (C18H20N2O4), Calcd (Found): C 58.99 (58.70), H 5.59 (5.23), N 7.77 (7.94).
3-Ethyl 5-Isopropyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6g)Recrystallization from methanol afforded (0.449 g, 70%): Rf=0.77 (CHCl3–MeOH, 95 : 5); mp: 132–134°C; 1H-NMR (400 MHz, CDCl3) δ: 1.10 (d, J=6.0 Hz, 3H, CH(CH3)2), 1.21 (d, J=7.2 Hz, 3H, CH(CH3)2), 1.26 (t, J=6.0 Hz, 3H, CH2CH3), 2.36 (s, 6H, C2-CH3, C6-CH3), 4.07–4.15 (m, J=3.6, 7.2 Hz, 2H, COOCH2), 4.93–5.00 (m, J=6.0, 6.4 Hz, 1H, CH(CH3)2), 5.11 (s, 1H, C4-H), 5.07 (s, 1H, NH), 7.38 (t, J=7.6, 8.0 Hz, 1H, aromatic C(5′)H), 7.65 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 8.02 (d, J=8.0 Hz, 1H, aromatic C(4′)H), 8.14 (s, 1H, aromatic C(2′)H); Anal. (%): (C20H24N2O6), Calcd (Found): C 61.84 (62.04), H 6.23 (5.94), N 7.21 (6.96).
3-(2-Methoxyethyl) 5-Methyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6h)Recrystallization from methanol afforded (0.432 g, 67%): Rf=0.52 (CHCl3–MeOH, 95 : 5); mp: 126–128°C; 1H-NMR (400 MHz, CDCl3) δ: 2.32 (s, 6H, C2-CH3, C6-CH3), 3.30 (s, 6H, OCH3 and COOCH3), 4.02–4.08 (m, J=4.0 Hz, 2H, COOCH2CH2O), 4.24–4.30 (m, J=4.0 Hz, 2H, COOCH2CH2O), 5.89 (s, 1H, C4-H), 5.98 (s, 1H, NH), 7.26 (t, J=8.0 Hz, 1H, aromatic C(5′)H), 7.48 (d, J=7.5 Hz, 1H, aromatic C(6′)H), 7.53 (d, J=8.0 Hz, 1H, aromatic C(4′)H), 7.75 (s, 1H, aromatic C(2′)H); Anal. (%): (C19H22N2O5), Calcd (Found): C 58.46 (58.32), H 5.68 (5.69), N 7.18 (7.11).
3-Ethyl 5-(2-Methoxyethyl) 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6i)Recrystallization from methanol afforded (0.487 g, 73%): Rf=0.56 (CHCl3–MeOH, 95 : 5); mp: 120–122°C; 1H-NMR (400 MHz, CDCl3) δ: 1.17 (t, J=4.0 Hz, 3H, CH2CH3), 2.31 (s, 6H, C2-CH3, C6-CH3), 3.33 (s, 3H,OCH3), 3.53–3.62 (m, J=4.0, 7.2 Hz, 2H, COOCH2CH2O), 4.02–4.12 (m, J=4.0, 6.4 Hz, 2H, CH2CH3), 4.16–4.27 (m, J=4.0, 6.4 Hz, 2H, COOCH2CH2O), 5.86 (s, 1H, C4-H), 7.26 (t, J=7.6, 8.0 Hz, 1H, aromatic C(5′)H), 7.48 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 7.53 (d, J=8.0 Hz, 1H, aromatic C(4′)H), 7.73 (s, 1H, aromatic C(2′)H); Anal. (%): (C20H24N2O7), Calcd (Found): C 59.40 (59.09), H 5.98 (5.85), N 6.93 (6.99).
3-Benzyl 5-Methyl 2,6-Dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate (6j)Recrystallization from methanol afforded (0.446 g, 64%): Rf=0.73 (CHCl3–MeOH, 95 : 5); mp: 114–116°C; 1H-NMR (400 MHz, CDCl3) δ: 2.26 (s, 3H, COOCH3), 3.55 (s, 6H, C2-CH3, C6-CH3), 3.59 (s, 2H, COOCH2), 5.09 (s, 1H, C4-H), 6.10 (s, 1H, NH), 7.07–7.27 (m, 11H, aromatic OCH2C6H5), 7.22 (t, J=7.6, 8.0 Hz, 1H, aromatic C(4′)H), 7.48 (d, J=7.6 Hz, 1H, aromatic C(6′)H), 7.89 (d, J=8.0 Hz, 1H, aromatic C(3′)H) 7.97 (s, 1H, aromatic C(2′)H); Anal. (%): (C23H22N2O6), Calcd (Found): C 65.39 (65.75), H 5.52 (5.10), N 6.63 (6.50).
Pharmacological EvaluationA written permission was obtained from the Research Ethics Committee (REC) of Faculty of Pharmacy, Tanta University and the National Research Center (NRC) where pharmacological evaluation was performed in accordance with Guide for the Care and Use of Laboratory Animals 8th Edition 2011 since Egypt has not yet compiled its own guide.
The intestine of male guinea-pigs weighing 300–450 g was removed above the ileocecal junction and longitudinal smooth muscle segments of 2 cm length were mounted under a resting tension of 0.5 g. The segments were maintained at 37°C in a 10 mL jacketed organ bath (ML870B6/C, PanLab). The organ bath was filled with a physiological saline solution of the following composition (mmol): NaCl (137), CaCl2 (1.8), KCl (2.7), MgSO4 (1.1), sodium hydride (NaH)2PO4 (0.4), NaHCO3 (12) and glucose (5), and was continuously aerated with carbogen (oxygen–carbon dioxide; 95 : 5) at 37°C. The muscles were equilibrated for 1 h with a solution changed every 15 min. The contractions were recorded with a force displacement transducer (MLT0201, PanLab) connected to amplifier. Stocks of the tested compounds (0.01 mM in ethanol) were stored protected from light. Dilutions were made with double distilled water.23)
In order to study the effects of synthesized DHP on KCl-induced contraction of ileum, at the first step, several contractions with KCl (40 mM) were made. No significant differences between KCl-induced contractions were observed as stability of tissue and thereafter the main experiments started. At this step, KCl (40 mM) elicited contraction was recorded again and the peak of the first phase (phasic contraction) was considered as a control. The contractile response was taken as the 100% value for the tonic (slow) component of the response. Then, tested compounds were cumulatively added and compound induced relaxation of contracted muscle was expressed as percent of control.
The IC50 of each compound (molar concentration needed to produce 50% relaxation on contracted ileal smooth muscle) was graphically determined by the concentration–response curves. Each segment was treated with only one compound. Nifedipine was used as a reference compound. A triplicate experiment was performed for each compound.
Data was statistically analyzed using SPSS 21.0 program (SPSS, Chicago, IL, U.S.A.).24) The results were recorded as the mean±standard error of the mean (S.E.M.) and they were evaluated statistically using one-way ANOVA following post hoc test.
Molecular DockingHerewith, we correspond to the situation where only the sequence of the target protein (and not the 3D structure) is available, and then its similarity with protein sequences in database(s) is analyzed. Consequently, suitable coordinates of the DHP receptor model were used.25) The Zhorov model was selected as the target receptor for a molecular docking study. All ligands were drawn into Marvin Sketch 5.11.4.26) The most energetically favored conformer was saved as (*. mol2) file format for docking. The optimal geometry of the ligands was determined during the docking process. The Molecular docking study was performed on selected molecules along with the reference molecule (nifedipine) into the DHPs receptor model active site using AutoDock Vina 1.1.2 protocol.27,28) Docking results were visualized using Discovery Studio 3.1 visualizer (Accelrys Software Inc., San Diego, CA, U.S.A.). The docking parameters are saved in a configuration file which is provided as an example in the supporting data.
3D-QSAR Studies Using Comparative Molecular Field Analysis (CoMFA) MethodCoMFA is an alignment-dependent descriptor method. Molecular field interaction energies are calculated and correlated with biological activities by using multivariate statistical analyses. In this research, we aim to generate a QSAR model based on 47 ligands with different core structures to generate a useful model for the prediction of novel ligands after. All ligands were drawn into Marvin Sketch 5.11.4. The most energetically favored conformer was saved as (*. mol2) file format by VAMPS module in BIOVIA Materials Studio 8.29) CoMFA has been carried out using SYBYL CERTARA X2.1.1.30)
The SYBYL engine consists of 2D and 3D studies: we selected the 3D studies to explain QSAR for the newly synthesized compounds. The molecules were aligned by the template based techniques using the basic nucleus. 3D descriptors such as Tripos force field, electrostatic, steric and hydrophobic field types, with cut-off values of 30.0 and 20.0 kcal/mol, respectively with Gasteiger charge type were selected for building the model.
The set of molecules was divided into training and test set based on the OptiSim Diversity Algorithm leading to a training set of 32 molecules which had been aligned was placed in a rectangular grid. The minus Log the half maximal inhibitory concentration (pIC50) values of DHPs were used as the dependent variables, CoMFA values as independent variable. A better value of q2 was obtained when hydrophobicity descriptors were used as an additional independent variable. Regression analyses of these variables were performed using the Partial least square (PLS) algorithms with default parameters. To acquire the optimal principal components (PCs) and q2 value for PLS procedure, the cross-validation analyses were performed by using leave-one-out (LOO) with 3 principal components. Both of the predicted pIC50 in the training set and the test set were obtained from the CoMFA model.
Though a high value of q2 is important, it alone is not sufficient for a predictive model. To determine the best CoMFA model, linear regressions were analyzed for the observed and predicted values of the test set compounds with intercept (squared correlation coefficient r2, slope k) and without intercept (squared correlation coefficient r02, slope k′). Models are considered acceptable if they meet all of the following conditions: 0.85≤k≤1.15 or 0.85≤k′≤1.15; q2>0.5, r2>0.6 and r2Pred>0.6.31)
Results and Discussion
ChemistryThe synthetic routes of the two DHP series which are based on Hantzsch condensation32) or a modified Hantzsch reaction are summarized in Chart 1. A series of symmetric achiral DHPs (3a–l) has been synthesized by using classical Hantzsch condensation.33) The asymmetric chiral DHPs series (6a–j) has been synthesized by a modified Hantzsch reaction as described by Meyer et al.,33) In spite the commonness of Hantzsch condensation, we still use due to its simplicity as well as the high yield of the product as a one-pot reaction. The synthesized compounds have either 2-nitrophenyl or 3-nitrophenyl ring at the 4-position of the DHP ring. All structural features necessary to reserve pharmacological activity were considered. Divergent acetoacetate esters were selected to give a variety of side chains which could help in building the 3D-QSAR model and the biological evaluation. Some acetoacetate is common and other is new such as the methoxy ethyl derivative. This type of selection provides a suitable range of bulkiness and electrostatic effects which is complied with SAR of DHPs.

Chart 1. Synthesis of Symmetrical Compounds (3a–l) and Asymmetrical Compounds (6a–j)
The preparation, collection and purification of the products were carried out in the absence of oxidizing agents and in darkness for enhanced photo-stability.34) The elemental analysis results for the synthesized compounds were within ±0.4% of the calculated values and the Proton-NMR spectra for the synthesized compounds in CDCl3 were compatible with the assigned structures. The structures of test compounds were shown in Table 1.
Table 1. Chemical Structures and the Results of
in Vitro Calcium Channel Antagonist Activity Presented as IC
50±S.E.M. of the Synthesized 1,4-Dihydropyridines; Symmetrical Compounds (
3a–
l) and Asymmetrical Compounds (
6a–
j)
| Compd. | R1 | R2 | X | IC50 (±S.E.M.)a) |
|---|
| Series 1 (3a–l) |
| 3a (Nifed.) | –CH3 | — | 2-NO2 | 6.32×10−10 (±0.035) |
| 3b | –C2H5 | — | 2-NO2 | 1.45×10−9 (±0.035) |
| 3c | –CH(CH3)2 | — | 2-NO2 | 1.60×10−9 (±0.041) |
| 3d | –CH2CH(CH3)2 | — | 2-NO2 | 1.44×10−9 (±0.173) |
| 3e | –CH2CH2OCH3 | — | 2-NO2 | 4.40×10−10(±0.091) |
| 3f | –CH2C6H5 | — | 2-NO2 | 3.07×10−9 (±0.058) |
| 3g | –CH3 | — | 3-NO2 | 2.60×10−9(±0.032) |
| 3h | –C2H5 | — | 3-NO2 | 2.10×10−9(±0.153) |
| 3i | –CH(CH3)2 | — | 3-NO2 | 2.88×10−9(±0.161) |
| 3j | –CH2CH(CH3)2 | — | 3-NO2 | 2.99×10−9(±0.066) |
| 3k | –CH2CH2OCH3 | — | 3-NO2 | 3.81×10−10(±0.002) |
| 3l | –CH2C6H5 | — | 3-NO2 | 2.83×10−9 (±0.058) |
| Series 2 (6a–j) |
| 6a | –C2H5 | –CH3 | 2-NO2 | 2.83×10−9 (±0.058) |
| 6b | –CH(CH3)2 | –C2H5 | 2-NO2 | 1.21×10−9 (±0.121) |
| 6c | –CH2CH2OCH3 | –CH3 | 2-NO2 | 9.6×10−10 (±0.261) |
| 6d | –CH2CH2OCH3 | –C2H5 | 2-NO2 | 1.61×10−9 (±0.133) |
| 6e | –CH2C6H5 | –CH3 | 2-NO2 | 2.35×10−9 (±0.113) |
| 6f | –C2H5 | –CH3 | 3-NO2 | 3.34×10−9 (±0.012) |
| 6g | –CH(CH3)2 | –C2H5 | 3-NO2 | 1.32×10−9 (±0.321) |
| 6h | –CH2CH2OCH3 | –CH3 | 3-NO2 | 2.40×10−9 (±0.118) |
| 6i | –CH2CH2OCH3 | –C2H5 | 3-NO2 | 1.78×10−9 (±0.104) |
| 6j | –CH2C6H5 | –CH3 | 3-NO2 | 2.48×10−9 (±0.089) |
a) The molar concentration of antagonist test compound causing a 50% in the tonic contractile response (IC50±S.E.M.) in guinea-pig ileum smooth muscle by KCl (80 mmol/L) was determined graphically by dose–response curve. Each compound is tested in triplicate.
The tested DHPs displayed varying levels of intrinsic calcium antagonist activity using high K+ contraction guinea-pig ileal longitudinal smooth muscle (GPILSM) method.35–39) The IC50 values ranged from 0.381 nM for 3k to 3.34 nM for 6f. The IC50 values were extracted from the graph using Graphpad Prism V6.01 software. A triplicate experiment was performed for each compound. The calculated S.E.M. reflected non-significant variation in the results. The results were presented as IC50±S.E.M. in Table 1. These results were tested statistically for significance using one-way ANOVA following post hoc test in SPSS 21 software. The results were statistically significant (p=0.000). This indicates that the obtained results were due to the intrinsic activity of each compound. The synthesized compounds showed activity similar to, higher or lower than the reference compound; nifedipine. The most active compound was 3k which is approximately two times more active than nifedipine.
A comprehensive study of the activity data shows that IC50 share a similar tendency suggesting that the hypertensive effect was produced by inhibitory action against L-type calcium channels. In the series of symmetric DHPs derivatives (3a–l), increasing lipophilic nature of R1 enhances the pharmacological activity to a certain limit. Therefore, 3f and l express high IC50 values proving our concept. Accordingly, the chloro-derivatives should express a higher pharmacological activity than the nitro-derivatives which is represented in Table 2.
Table 2. Actual and Predicted Biological Activities
| Code | IC50 (nM) (Obs.) | PIC50 (Obs.) | PIC50 (Pred.) | Residues | c Log P |
|---|
| Training Set from Nitro Analogues (17 ligands) |
| 3c | 1.60 | 8.7959 | 8.902 | −0.1061 | 4.8008 |
| 3d | 1.45 | 8.386 | 8.854 | −0.468 | 6.0388 |
| 3e | 0.44 | 9.3565 | 9.149 | 0.2075 | 3.0448 |
| 3g | 2.60 | 8.585 | 8.6975 | −0.1125 | 3.2048 |
| 3h | 2.10 | 8.6778 | 8.7505 | −0.0727 | 4.2628 |
| 3i | 2.88 | 8.5406 | 8.7257 | −0.1851 | 4.8808 |
| 3j | 2.99 | 8.5406 | 8.551 | −0.0104 | 6.1188 |
| 6a | 1.21 | 8.9172 | 8.9219 | −0.0047 | 3.6538 |
| 6b | 0.96 | 9.0177 | 8.7946 | 0.2231 | 4.4918 |
| 6c | 1.61 | 8.7932 | 8.8299 | −0.0367 | 3.0848 |
| 6d | 2.35 | 8.6289 | 8.6141 | 0.0148 | 3.6138 |
| 6e | 3.34 | 8.4763 | 8.3474 | 0.1289 | 1.8884 |
| 6f | 1.32 | 8.8794 | 8.7561 | 0.1233 | 3.7338 |
| 6g | 2.40 | 8.6198 | 8.6829 | −0.0631 | 4.5718 |
| 6h | 2.87 | 8.5421 | 8.7278 | −0.1857 | 3.1648 |
| 6i | 1.72 | 8.7645 | 8.6644 | 0.1001 | 3.6938 |
| 6j | 2.49 | 8.6038 | 8.5177 | 0.0861 | 4.9234 |
| Training Set from Chloro Analogues (15 ligands) |
| 5a* | 0.43 | 9.3665 | 9.5697 | −0.2032 | 4.1442 |
| 5b | 0.23 | 9.4559 | 9.4517 | 0.0042 | 4.7038 |
| 5c | 0.35 | 9.4559 | 9.4037 | 0.0522 | 4.7082 |
| 5d | 0.48 | 9.3118 | 9.4483 | −0.1365 | 5.5418 |
| 5e | 0.77 | 9.1135 | 9.0374 | 0.0761 | 5.2372 |
| 5f | 0.59 | 9.2291 | 9.1732 | 0.0559 | 6.1608 |
| 5g | 0.27 | 9.5686 | 9.5054 | 0.0632 | 4.1348 |
| 5h | 0.72 | 9.1427 | 9.4504 | −0.3077 | 4.6638 |
| 5i | 0.70 | 9.1549 | 9.0852 | 0.0697 | 5.9686 |
| 5j | 0.60 | 9.2218 | 9.3811 | −0.1593 | 4.3992 |
| 5k | 0.28 | 9.5528 | 9.4067 | 0.1461 | 5.2286 |
| 5l | 0.27 | 9.5686 | 9.4302 | 0.1384 | 5.2372 |
| 5m | 0.6 | 9.2218 | 9.2821 | −0.0603 | 5.8476 |
| 5n | 0.75 | 9.1249 | 9.4068 | −0.2819 | 4.2674 |
| 5o | 0.26 | 9.5850 | 9.5182 | 0.0668 | 4.3592 |
| Test Set from Nitro Analogues (5 ligands) |
| Nifed | 0.63 | 9.2007 | 9.1709 | 0.0298 | 3.1248 |
| 3b | 1.45 | 8.8386 | 8.7666 | 0.072 | 4.1828 |
| 3f | 3.07 | 8.5129 | 8.4758 | 0.0371 | 6.274 |
| 3k | 0.38 | 9.4202 | 9.3607 | 0.0595 | 3.1248 |
| 3l | 2.83 | 8.5482 | 8.5806 | −0.0324 | 6.642 |
| Test Set from Chloro Analogues (10 ligands) |
| 4a* | 0.42 | 9.3768 | 9.412 | −0.0352 | 3.8702 |
| 4b | 0.20 | 9.699 | 9.4485 | 0.2505 | 5.2328 |
| 4c | 0.40 | 9.3979 | 9.5076 | −0.1097 | 5.8508 |
| 4d | 0.54 | 9.2676 | 9.2622 | 0.0054 | 7.0546 |
| 4e | 0.20 | 9.699 | 9.5725 | 0.1265 | 4.0948 |
| 4f | 0.86 | 9.0655 | 8.9548 | 0.1107 | 7.612 |
| 4g | 0.48 | 9.3188 | 9.3672 | −0.0484 | 4.3906 |
| 4h | 0.16 | 9.7959 | 9.3986 | 0.3973 | 4.9282 |
| 4i | 0.41 | 9.3872 | 9.3185 | 0.0687 | 7.3046 |
| 4j | 0.70 | 9.1549 | 9.4923 | −0.3374 | 6.0666 |
* The chemical structures of the chloro-derivatives are provided in the supporting data.
Computational molecular docking is widely used for the study of protein–ligand interactions and for drug discovery and development. Typically, the process starts with a target of known structure, such as a crystallographic structure of an enzyme of medicinal interest or a homology model. Docking is then used to predict the bound conformation and the binding free energy of small molecules to the target. Single docking experiments are useful for exploring the function of the target, and virtual screening- in which a large library of compounds is docked and ranked-may be used to identify new inhibitors for drug development.
To our knowledge, several theoretical models of CaV1.2-DHPs have been reported. In 2001, Zhorov et al. built two models of the pore region of CaV1.2 from rabbit cardiac muscle, and docked nifedipine into the active site to explore the interactions of agonists and antagonists.40) In 2003, Lipkind and Fozzard constructed the inner pore structure of CaV1.2 and predicted the binding conformations of nifedipine, phenylalkylamine and agonist Bay K8644 by using the molecular docking approach.41) In 2007, Cosconati et al. constructed the central pore of CaV1.2 and explored the binding modes of nine different DHPs.42) In 2009, Tikhonov and Zhorov constructed two structural models of CaV1.2 in open and closed states, and explored the possible binding structures of (S)-nimodipine.43)
These computational results provided some valuable information on L-type Ca2+ channels (LTCC)-DHP recognition, but the reliability of the predictions may be questionable because the template used in the homology modeling of CaV1.2 is the K+ channel, where the structures of VGCC and the K+ channel are quite different, especially in the intervening P-loop.2)
Recently, the crystal structure of the voltage-gated calcium channel from bacterium Arcobacter butzleri (CaVAb) was reported (PDB entry: 4MS2), with a pore-motif that is structurally related to vertebrate VGCCs. The residues forming the inner low-affinity site of CaVAb are still present in vertebrate channels. Therefore, construction of the 3D structure of human CaV1.2 based on the crystal structure of CaVAb is more reasonable than that of the K+ channel.44)
In the current study, we performed pre-synthetic docking of 21 DHPs derivatives into the active site of Zhorov model using AutoDock Vina 1.1.2 protocol. The crystal structure of Zhorov model was obtained from the scientist himself with a permission of a scientific use. Besides, we are working now on a homology model based on bacterium Arcobacter butzleri (CaAb) and the docking results will be compared and published in the recent future. In the meantime, the overall architecture of the mammalian skeletal muscle CaV1.1 channel was recently elucidated at a resolution of ca. 4–6 Å by cryo-electron microscopy.44) However, a higher-resolution structural analysis of mammalian CaV channels has not yet been achieved.
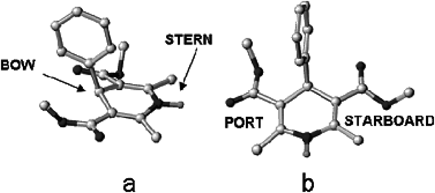
Hereafter, to distinguish between the two sides of the DHP ring, as suggested by Goldmann and Stoltefuss,45) the preferred conformation of the DHP ring will be regarded as a flattened boat with C4 as the bow, the axial aryl ring as the bowsprit, and the N1 atom as the stern (Fig. 1a). Accordingly, the two sides of the DHP ring will be referred to the port side (left) and the starboard side (right; Fig. 1b).
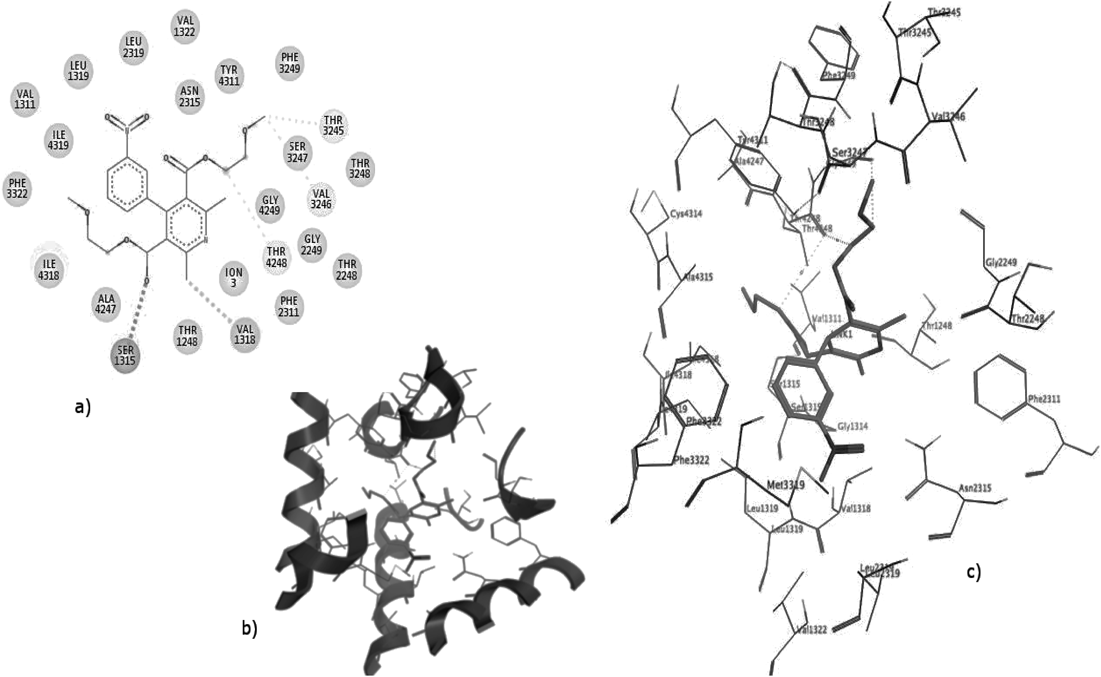
Based on our previous studies,46) the best binding poses of nifedipine showed that NH group of the DHP ring makes H-bond with Tyr4311 in the stern-to-Tyr4311 orientation. This was proved by the docking scores, and the surrounding residues of ligands, especially the residues that have essential impact on the binding affinity between LTCC and DHPs (Table 3).
Table 3. The Surrounding Residues of the Best Docking Poses of DHPs Binding in the Active Site of LTCC Are Shown Below, and Experimentally Found DHP-Sensing Residues Are in Bold Type
| Ligand | Surrounding residues | Docking score (kcal/mol) |
|---|
| Nifed. | Ile4318, Val1322, Thr3245, Thr3248, Phe3247, Ser3247, Thr4248, Ile3311, Met3319, Ile3314, Ile4315, Met3318, Ala3315, Phe3322, Tyr4311. | −7.0 |
| 3k | Ile4318, Val1322, Thr3245, Thr3248, Phe3247, Ser3247, Thr4248, Ile3311, Met3319, Ile3314, Ile4315, Met3318, Ala3315, Phe3322, Tyr4311. | −8.4 |
| 3f | Ile4318, Val1322, Thr3245, Thr3248, Phe3247, Ser3247, Thr4248, Ile3311, Met3319, Ile3314, Ile4315, Met3318, Ala3315, Phe3322, Tyr4311. | −6.6 |
It can be observed in Fig. 2, that the dihydropyridine ring of DHPs fits into the cleft formed by the IIIS6, IIIS5 and IVS6 segments, and the plane of the DHP ring is parallel to the pore axis, while the 4-aryl substituent is perpendicular to the pore axis.45) The starboard side of the heterocyclic ring faces upward, and the portside side at the ortho position to the NH group faces towards the selectivity filter. Most of the ligands fit into a hydrophobic pocket surrounded by Phe2322, Ile1150, Phe1176, Met3318 and Met3319, and form aryl–aryl interaction with Phe3322 or Tyr4311. The nitro group and oxygen of the carbonyl group in nifedipine chelate the Ca2+ cofactor of the selectivity filter.
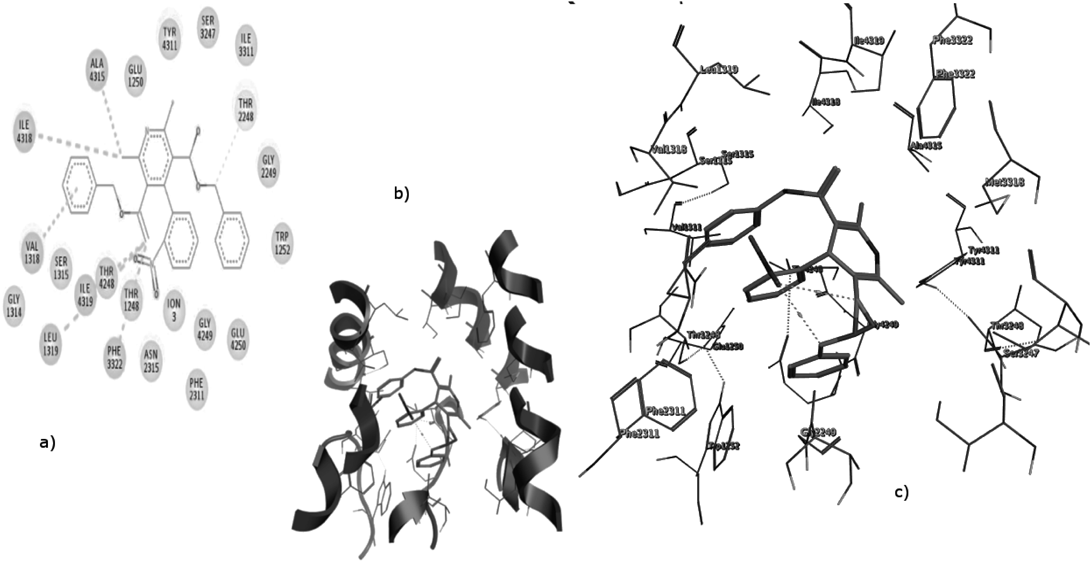
In Fig. 3, the bowsprit group of 3k points upward and then approaches the Ca2+ ion chelated by the acid residues of the selectivity filter, while the bowsprit group of nifedipine cannot due to the side chain length privilege in case of 3k. In addition, the portside group of 3k is closer to the selectivity filter in comparison with that of nifedipine. These results are consistent with previous studies that the antagonistic action of DHP derivatives is directly associated with the Ca2+ occupancy of the selectivity filter.
The steric effect of the 2 phenyl rings pushes the crucial Tyr4311 away from NH of DHP ring also the nitro and carbonyl groups that chelate Ca2+ ion not directed to the lumen of the selectivity filter. This could show the inability of 3f to undergo well allosteric inhibition (Fig. 4).
As a conclusion, we can understand the best binding mode of DHPs in the active site by molecular alignment of highly active ligands (Fig. 5) to describe the amino acid interactions and be correlated with docking scores shown in Table 3.
3D-QSAR Studies Using CoMFA 3D-QSAR MethodIn order to give a systematic evaluation on DHPs as CCB and to explore more potent inhibitors, 3D-QSAR models were built using the 3D-QSAR protocol of SYBYL CERTARA® X2.1.1. In this study, 47 compounds bearing variable cores either nitro derivatives or chloro derivatives46) with definite IC50 values were selected as the model dataset. By convention, the pIC50 scale (−log IC50), in which higher values indicate exponentially greater potency, is used as a method to measure inhibitory activity. The training and test set was chosen by the OptiSim Diversity Algorithm in SYBYL. The diversity algorithm resulted in a perfect selection for both training and test sets. The training set contains 32 ligands with varieties of ortho and meta substituents on the phenyl ring and diverse side chains which could give a reliable data for QSAR model away from over fit of data. The test set contains 15 ligands. 3k (the highest biologically active compound in the nitro series) is selected in concordance with 3e selection in the training set to reflect the reliability of the basis of diversity algorithms.
Molecular AlignmentOne of the most crucial problems in most of the alignment-based 3D-QSAR methods is that their results are highly sensitive to the manner in which the bioactive conformations of all the molecules are superimposed over each other. In cases, where all the molecules in a data set have a common rigid core structure, molecules can be aligned easily using least-square fitting procedure. However, in case of structural heterogeneity in the dataset, alignment of highly flexible molecules becomes quite difficult and time consuming.
In CoMFA studies, 3D structures of the molecules are required to be aligned based on a suitable conformational template and its substructure, which is assumed to be a “bioactive” conformation. A compound with the highest pIC50 value was used as the template for molecular alignment. Three alignment figures are shown to describe the nitro-analogues series, chloro-analogues series and all 47 ligands using the SYBYL align database methods (Fig. 6).
Model ValidationTo seek the most robust QSAR model, different sets of molecular descriptors either 2D or 3D have been chosen. The results of each model have been presented in Table 4. According to the previous data, model 3 has been chosen due to the relatively high (Q2) and (R2) values. Better values of R2 and Q2 were obtained when the calculated logarithm of the 1-octanol–water partition coefficient of the non-ionized molecules (c Log P) was used as an additional independent variable that signifies the need of hydrophobicity for high biological activity.
Table 4. Different QSAR Models with the Cross-Validated Coefficient (
Q2) and Correlation Coefficient (
R2)
| Model | Molecular descriptors | (Q2) | (R2) | Standard error |
|---|
| Model 1 | CoMFA | 0.641 | 0.840 | 0.161366 |
| Model 2 | c Log P*, CoMFA | 0.660 | 0.847 | 0.159536 |
| Model 3 | c Log P, CoMFA, Hydrophobe | 0.686 | 0.939 | 0.117327 |
In Table 5, we present the data of our QSAR model and PLS results to prove its reliability and predictability according to the validation criteria of Golbraikh and Tropsha47) (Fig. 7).
Table 5. Validation Criteria for Our QSAR Model
| R2 | Q2 | R2pred | r0 | r0′ |
|---|
| 0.939 | 0.686 | 0.893 | 0.9111 | 0.902 |
| K | K′ | S.E. | RMSE | |
| 0.897 | 0.968 | 0.1173277 | 0.025284 | |
In the CoMFA contour maps, the steric field is illustrated in green (bulky favorable) and yellow (bulky unfavorable) contours (Fig. 8-left). The steric favorable green contours near the phenyl ring, C-3 and C-5 substituents of the DHP ring indicated the importance of bulky groups in these areas for inhibitory activity. The presence of bulky hydrophilic groups on the phenyl ring increased the inhibitory activity in comparing 4e and 5e which is proved by large % Steric/Hydrophobic desirable contribution 27.17 and 34.12%, respectively. 3k shows a less percent (6.35%) compared to chloro derivatives but higher than nifedipine (5.13%).
One of steric unfavorable yellow contours was near C-2 and C-4 dimethyl groups of the DHP ring (Fig. 8-right), suggesting that a bulky group in this region decreased inhibitory activity as illustrated by the fact that pIC50 of 3k is higher than 3l and f which have benzyl group in the yellow region causing a penalty and rationalize the high % Steric/Hydrophobic undesirable contribution of them (7.89 and 9.86%, respectively (Graphical abstract contains the colored image).
In the CoMFA electrostatic contour map (Fig. 8), red contours near the substituent on the phenyl ring and the carbonyl groups of the ester linkages, indicated that electronegative groups around these areas increased the blocking activity of DHPs. With an electronegative substituent, such as nitro groups and extra oxygen atom in the side chain of 3k rationalize the large percent of electrostatic negative charge desirable (62.65%) than nifedipine (65.59%). In case of 4e and 5e, the lower % of contribution favors the nitro substitutions.