Experimental
GeneralUV spectra were recorded on a JASCO V-650 instrument. IR spectra were measured on a Perkin-Elmer spectrum 100 Fourier transform (FT)-IR spectrometer. 1H- and 13C-NMR spectra were determined on a Varian Gemini-300 superconducting FT-NMR spectrometer. The chemical sifts are expressed in ppm relative to tetramethylsilane (δ=0) as internal standard for 1H-NMR and CDCl3 (δ=77.0) for 13C-NMR. Mass spectra were taken on a Thermo Fisher Scientific Exactive spectrometer. Optical rotations were measured on a JASCO P-2200 polarimeter.
Flash column chromatography (CC) was performed on using Kanto Silica Gel 60N. HPLC analyses were performed on GL Sciences-GL7400 instrument with a photodiode array detector.
All operations were carried out under nitrogen or argon. Evaporation of the extract or the filtrate was carried out under reduced pressure. In solvent extraction procedure, organic layer was dried over anhydrous Na2SO4. Ether refers to diethyl ether, and hexane to n-hexane.
(E)-4-[(R)-4-Hydroxy-2,6,6-trimethylcyclohex-1-enyl]but-3-en-2-one (2)HF·Pyridine (Py) (4 mL) was added in several portions to a solution of compound 115) (987 mg, 3.06 mmol) in tetrahydrofuran (THF) (10 mL) at 0°C. After being stirred at 0°C for 40 min, the reaction mixture was diluted with AcOEt, washed successively with brine, saturated aqueous NaHCO3, and then brine. Evaporation of the dried solution gave a residue, which was purified by flash CC (acetone–hexane, 3 : 7) to afford compound 2 (638 mg, quant.) as a coreless oil: [α]D23 −87.8 (c=1.01, CHCl3). IR (CHCl3) cm−1: 3606 and 3460 (OH), 1668 (CO), 1605 (C=C). UV λmax (MeOH) nm (ε): 291 (9560). 1H-NMR (300 MHz, CDCl3) δ: 1.11 and 1.12 (each 3H, s, gem-CH3), 1.49 (1H, t, J=12 Hz, 5′-H), 1.71 (1H, d, J=4.5 Hz, OH), 1.77 (3H, s, 2′-CH3), 1.78 (1H, ddd, J=12, 3.5, 2 Hz, 5′-H), 2.09 (1H, br dd, J=17, 9.5 Hz, 3′-H), 2.30 (3H, s, CH3CO), 2.43 (1H, br dd, J=17, 5.5 Hz, 3′-H), 4.00 (1H, m, 4′-H), 6.10 (1H, d, J=16.5 Hz, 3-H), 7.19 (1H, br d, J=16.5 Hz, 4-H). 13C-NMR (75 MHz, CDCl3) δ: 21.55, 27.27, 28.53, 30.04, 36.87, 42.73, 48.37, 64.45, 132.28, 132.32, 135.58, 142.33, 198.55. High resolution (HR)-MS (electrospray ionization (ESI)) m/z Calcd for C13H20O2Na [M+Na]+ 231.1356. Found 231.1354.
(R)-3,5,5-Trimethyl-4-[(E)-3-oxobut-1-enyl]cyclohex-3-enyl 3,5-Dinitrobenzoate (3)To a stirred solution of compound 2 (1.43 g, 6.87 mmol), Et3N (1.44 mL, 10.4 mmol) and DMAP (42 mg, 0.34 mmol) in dry CH2Cl2 (35 mL) was added 3,5-dinitrobenzoyl chloride (1.59 g, 6.89 mmol) at 0°C and the mixture was stirred at 0°C for a further 60 min. After the reaction was quenched by addition of saturated aqueous NH4Cl, CH2Cl2 was evaporated off. The resulting mixture was diluted with AcOEt and washed with brine. The organic layer was dried and evaporated to give a residue, which was purified by flash CC (AcOEt–hexane–CH2Cl2, 1 : 5 : 3) to provide compound 3 (2.48 g, 90%) as a colorless solid: mp 102–105°C. [α]D23 −56.9 (c=1.00, CHCl3). IR (CHCl3) cm−1: 1729 and 1670 (CO), 1628 and 1607 (C=C), 1549 and 1345 (NO2). UV λmax (MeOH) nm (ε): 209 (37000), 285 (11300). 1H-NMR (300 MHz, CDCl3) δ: 1.19 and 1.24 (each 3H, s, gem-CH3), 1.83 (3H, s, 3-CH3), 1.84 (1H, t, J=12 Hz, 6-H), 2.02 (1H, ddd, J=12, 4, 2 Hz, 6-H), 2.34 (3H, s, CH3CO), 2.40 (1H, br dd, J=17.5, 10 Hz, 2-H), 2.68 (1H, br dd, J=17.5, 5.5 Hz, 2-H), 5.42 (1H, m, 1-H), 6.16 (1H, d, J=16.5 Hz, 2′-H), 7.22 (1H, br d, J=16.5 Hz, 1′-H), 9.16 (2H, d, J=2.5 Hz, ArH), 9.24 (1H, t, J=2.5 Hz, ArH). 13C-NMR (75 MHz, CDCl3) δ: 21.41, 27.46, 28.35, 29.85, 36.72, 38.42, 43.85, 71.07, 122.33, 129.35 (C×2), 130.36, 132.89, 134.19, 136.01, 141.47, 148.65 (C×2), 162.07, 198.22. HR-MS (ESI) m/z Calcd for C20H22O7N2Na [M+Na]+ 425.1319. Found 425.1320.
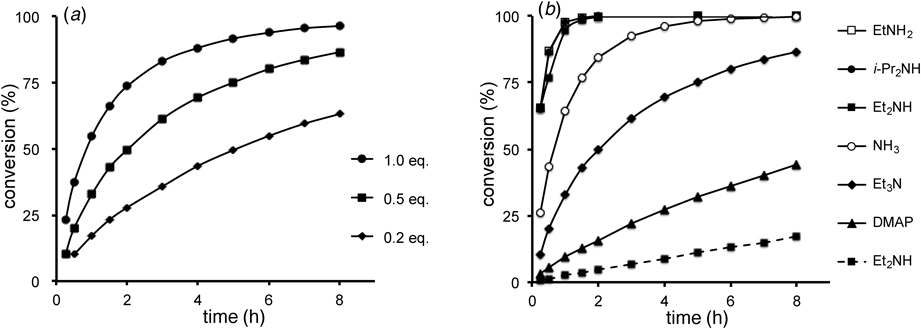
Time Course of Alcoholysis of 3,5-Dinitrobenzoate 3 (Fig. 1)Each amine (1.0 M in MeOH; 20 µL, 0.02 mmol to 100 µL, 0.1 mmol) was added to a stirred solution of compound 3 (40.0 mg, 0.1 mmol) in MeOH or EtOH (5.0 mL). The reaction was monitored by HPLC [LiChrosorb CN 0.46×25 cm (Merck), AcOEt–hexane (3 : 7), 1.0 mL/min, 254 nm (detect.); methyl and ethyl 3,5-dinitrobenzoate: 4.0 min, 3: 5.1 min, 2: 6.1 min]. Conversions (%) of compound 3 into compound 2 were calculated as shown in the following equation.
Et2NH-Promoted Methanolysis of 3,5-Dinitrobenzoate 3To a stirred solution of compound 3 (402 mg, 1.00 mmol) in THF (1 mL) and MeOH (30 mL) was added Et2NH (1.0 M in MeOH; 0.5 mL, 0.5 mmol) at room temperature (r.t.). After being stirred at r.t. for 1 h, MeOH was evaporated off. Ether (10 mL) and hexane (10 mL) were added to the resulting residue and the precipitates were filtered off. The filtrate was concentrated to give a residue, which was purified by flash CC (acetone–hexane–CH2Cl2, 1 : 2 : 2) to yield compound 2 (208 mg, quant.) as a colorless oil.
(R)-3,5,5-Trimethyl-4-[(E)-3-oxobut-1-en-1-yl]cyclohex-3-enyl 2-Chloroacetate (4)To a stirred solution of compound 2 (823 mg, 3.95 mmol), Et3N (0.82 mL, 5.89 mmol) and DMAP (24 mg, 0.20 mmol) in dry CH2Cl2 (15 mL) was added slowly chloroacetyl chloride (0.38 mL, 4.78 mmol) at 0°C. After being stirred at 0°C for 1h, the mixutre was quenched by addition of saturated aqueous NH4Cl and extracted with AcOEt. The extracts were washed with brine, dried and evaporated to give a residue, which was purified by flash CC (AcOEt–hexane, 1 : 2) to provide compound 4 (825 mg, 73%) as a colorless solid: mp 75–77°C. [α]D20 −60.9 (c=0.98, CHCl3). IR (CHCl3) cm−1: 1749, 1687sh and 1669 (CO), 1607 (C=C). UV λmax (MeOH) nm (ε): 218 (7800), 287 (9700). 1H-NMR (300 MHz, CDCl3) δ: 1.12 and 1.16 (each 3H, s, gem-CH3), 1.65 (1H, t, J=12 Hz, 6-H), 1.77 (3H, s, 3-CH3), 1.85 (1H, ddd, J=12, 3.5, 2 Hz, 6-H), 2.21 (1H, br dd, J=17, 9.5 Hz, 2-H), 2.31 (3H, s, CH2CO), 2.54 (1H, br dd, J=17, 5.5 Hz, 2-H), 5.15 (1H, m, 1-H), 6.12 (1H, d, J=16.5 Hz, 2′-H), 7.19 (1H, br d, J=16.5 Hz, 1′-H). 13C-NMR (75 MHz, CDCl3) δ: 21.40, 27.42, 28.30, 29.76, 36.49, 38.28, 41.09, 43.58, 70.04, 130.76, 132.68, 135.75, 141.70, 166.93, 198.38. HR-MS (ESI) m/z Calcd for C15H22O3Cl [M+H]+ 285.1252 and 287.1223. Found 285.1252 and 287.1221.
Time Course of Et3N-Promoted Methanolysis of Chloroacetate 4 (Table 1)In the same manner as described for time course of alcoholysis of 3,5-dinitrobenzoate 3, a solution of compound 4 (28.5 mg, 0.1 mmol) in MeOH (5.0 mL) was treated with Et3N. The reaction was monitored by HPLC [LiChrosorb CN 0.46×25 cm (Merck), AcOEt–hexane (3 : 7), 1.0 mL/min, 254 nm (detect.); 4 (ε at 254 nm: 3080): 4.2 min, 2 (ε at 254 nm: 2550): 6.1 min].
(E)-3-[(R)-4-tert-Butyldimethylsilyloxy(TBS)-2,6,6-trimethylcyclohex-1-enyl]allyl Acetate (6)To a solution of compound 515) (1.50 g, 4.43 mmol) in dry ether (30 mL) was added diisobutylaluminium hydride (DIBAL-H) (1.0 M in hexane; 9.8 mL, 9.8 mmol) at 0°C and the mixture was stirred at 0°C for 10 min. Excess DIBAL-H was destroyed by the addition of moist silica gel (SiO2–H2O, 5 : 1) and the mixture was filtered through Celite. The filtrate was dried and evaporated to give the crude alcohol, which was dissolved in dry CH2Cl2 (10 mL) and Et3N (3.1 mL, 22.3 mmol), DMAP (54 mg, 0.44 mmol) and Ac2O (0.84 mL, 8.89 mmol) were added to it. After being stirred at r.t. for 20 min, the reaction mixture was diluted with AcOEt and washed with brine. The organic layer was dried and evaporated to give a residue, which was purified by flash CC (AcOEt–hexane, 15 : 85) to give compound 6 (1.31 g, 84% from 5) as a colorless oil: [α]D25 −77.5 (c=0.94, CHCl3). IR (CHCl3) cm−1: 1733 (CO). 1H-NMR (300 MHz, DCl3) δ: 0.78 (6H, s, SiCH3×2), 0.90 (9H, s, tert-Bu), 1.01 and 1.03 (each 3H, s, gem-CH3), 1.47 (1H, t, J=12 Hz, 5′-H), 1.65 (1H, ddd, J=12, 3.5, 2 Hz, 5′-H), 1.67 (3H, s, 2′-CH3), 2.04 (1H, br dd, J=17, 9.5 Hz, 3′-H), 2.07 (3H, s, CH3COO), 2.20 (1H, br dd, J=17, 5.5 Hz, 3′-H), 3.93 (1H, m, 4′-H), 4.61 (2H, dd, J=6.5, 1 Hz, 1-H2), 5.52 (1H, tt, J=16, 6.5 Hz, 2-H), 6.13 (1H, br d, J=16 Hz, 3-H). 13C-NMR (75 MHz, CDCl3) δ: −4.60 (C×2), 18.21, 21.00, 21.20, 25.91 (C×3), 28.25, 29.93, 36.64, 42.71, 48.56, 65.44, 65.52, 127.29, 127.65, 132.15, 136.02, 170.84. HR-MS (ESI) m/z Calcd for C20H36O3NaSi [M+Na]+ 375.2326. Found 375.2331.
(E)-3-[(R)-4-Hydroxy-2,6,6-trimethylcyclohex-1-enyl]allyl Acetate (7)In the same manner as described for the preparation of compound 2, desilylation of compound 6 (1.20 g, 3.40 mmol) gave crude products, which were purified by flash CC (acetone–hexane, 3 : 7) to provide compound 7 (677 mg, 83%) as a colorless oil: [α]D24 −116.3 (c=1.06, CHCl3). IR (CHCl3) cm−1: 3606 and 3453 (OH), 1732 (CO). 1H-NMR (300 MHz, CDCl3) δ: 1.03 and 1.04 (each 3H, s, gem-CH3), 1.44 (1H, t, J=12 Hz, 5′-H), 1.46 (1H, br s, OH), 1.68 (3H, s, 2′-CH3), 1.75 (1H, ddd, J=12, 3.5, 2 Hz, 5′-H), 2.00 (1H, br dd, J=17, 9.5 Hz, 3′-H), 2.07 (3H, s, CH3COO), 2.34 (1H, br dd, J=17, 5.5 Hz, 3′-H), 3.97 (1H, m, 4′-H), 4.60 (2H, dd, J=6.5, 1 Hz, 1-H2), 5.51 (1H, dt, J=16, 6.5 Hz, 2-H), 6.11 (1H, br d, J=16 Hz, 3-H). 13C-NMR (75 MHz, CDCl3) δ: 21.00, 21.18, 28.36, 29.95, 36.73, 42.17, 48.19, 64.96, 65.38, 126.64, 127.92, 131.94, 136.30, 170.87. HR-MS (ESI) m/z Calcd for C14H22O3Na [M+Na]+ 261.1461. Found 261.1463.
(R)-4-[(E)-3-Acetoxyprop-1-enyl]-3,5,5-trimethylcyclohex-3-enyl 3,5-Dinitrobenzoate (8)In the same manner as described for the preparation of compound 3, benzoylation of compound 7 (614 mg, 2.58 mmol) gave crude products, which were purified by flash CC (AcOEt–hexane–CH2Cl2, 1 : 5 : 3) to provide compound 8 (1.12 g, quant.) as a pale yellow solid: mp 77–80°C. [α]D24 −71.5 (c=1.02, CHCl3). IR (CHCl3) cm−1: 1729 (CO), 1548 and 1345 (NO2). 1H-NMR (300 MHz, CDCl3) δ: 1.12 and 1.17 (each 3H, s, gem-CH3), 1.75 (3H, s, 3-CH3), 1.80 (1H, t, J=12 Hz, 6-H), 1.97 (1H, ddd, J=12, 3.5, 1.5 Hz, 6-H), 2.10 (3H, s, CH3COO), 2.34 (1H, br dd, J=16.5, 9.5 Hz, 2-H), 2.59 (1H, br dd, J=16.5, 6 Hz, 2-H), 4.65 (2H, d, J=6.5 Hz, 3′-H2), 5.41 (1H, m, 1-H), 5.59 (1H, dt, J=16, 6.5 Hz, 2′-H), 6.16 (1H, br d, J=16 Hz, 1′-H), 9.16 (2H, d, J=2 Hz, ArH), 9.23 (1H, t, J=2 Hz, ArH). 13C-NMR (75 MHz, CDCl3) δ: 20.94, 21.04, 28.12, 29.69, 36.54, 37.84, 43.63, 65.13, 71.63, 122.22, 125.46, 128.56, 129.33 (C×2), 131.16, 134.34, 136.71, 148.59 (C×2), 162.08, 170.77. HR-MS (ESI) m/z Calcd for C21H24O8N2Na [M+Na]+ 455.1425. Found 455.1427.
Et2NH-Promoted Methanolysis of 3,5-Dinitrobenzoate 8In the same manner as described for Et2NH-promoted methanolysis of 3,5-dinitrobenzoate 3, methanolysis of compound 8 (200 mg, 0.46 mmol) gave the crude products, which were purified by flash CC (acetone–hexane–CH2Cl2, 2 : 9 : 9) to yield compound 2 (98 mg, 89%) as a colorless oil.
(S)-4-Ethynyl-3,5,5-trimethyl-2-oxocyclohex-3-enyl 3,5-Dinitrobenzoate (10)In the same manner as described for the preparation of compound 3, benzoylation of compound 9 (1.23 g, 6.90 mmol) gave crude products, which were purified by flash CC (AcOEt–hexane, 3 : 7) to provide compound 10 (2.21 g, 86%) as a colorless solid: mp 133–134°C. [α]D25 −166.3 (c=0.93, CHCl3). IR (CHCl3) cm−1: 3302 (≡C−H), 2092 (C≡C), 1742 and 1689 (CO), 1628 (C=C), 1549 and 1345 (NO2). 1H-NMR (300 MHz, CDCl3) δ: 1.41 and 1.47 (each 3H, s, gem-CH3), 2.03 (3H, s, 3-CH3), 2.27 (1H, dd, J=13, 7 Hz, 6-H), 2.33 (1H, t, J=13 Hz, 6-H), 3.88 (1H, s, ≡C–H), 5.82 (1H, dd, J=13, 7 Hz, 1-H), 9.20 (2H, d, J=2.5 Hz, ArH), 9.25 (1H, t, J=2.5 Hz, ArH). 13C-NMR (75 MHz, CDCl3) δ: 14.37, 25.91, 30.48, 36.87, 41.20, 73.24, 79.66, 93.37, 122.59, 129.68 (C×2), 133.44, 137.44, 145.19, 148.63 (C×2), 161.87, 191.79. HR-MS (ESI) m/z Calcd for C18H16O7N2Na [M+Na]+ 395.0850. Found 395.0850.
Et2NH-Promoted Methanolysis of 3,5-Dinitrobenzoate 10In the same manner as described for Et2NH-promoted methanolysis of 3,5-dinitrobenzoate 3, methanolysis of compound 10 (372 mg, 1.00 mmol) gave the crude products, which were purified by flash CC (acetone–hexane–CH2Cl2, 1 : 6 : 3) to yield compound 9 (176 mg, 99%) as a colorless solid. 1H-NMR data were identical with those previously reported.17) The enantiomeric purity was confirmed (>99% ee) by chiral HPLC [CHIRALPAK IC 0.46×25 cm (Daicel), EtOH–hexane (5 : 95), 1.0 mL/min, 27°C, 267 nm (detect.); 9: 11.13 min, enantiomer of 9: 12.69 min]; HR-MS (ESI) m/z Calcd for C11H14O2Na [M+Na]+ 201.0886. Found 201.0885.
(S)-3-Hydroxy-3,7-dimethyl-2-oxooct-6-enyl 3,5-Dinitrobenzoate (12a)To a solution of the compound 11a8) (1.00 g, 2.62 mmol) in dry THF (7.7 mL) and dry dimethylsulfoxide (7.7 mL) was added IBX (2.20 g, 7.86 mmol) in one portion and the reaction mixture was heated at 50°C for 1 h under stirring. After cooling, water (15 mL) was added to the mixture and the precipitates were filtered off and washed with AcOEt. The filtrate was washed with brine, dried and evaporated to give a residue, which was purified by flash CC (AcOEt–hexane, 3 : 7) to yield the ketone 12a (908 mg, 91%) as a pale yellow viscous oil: [α]D22 −23.1 (c=0.99, CHCl3). IR (CHCl3) cm−1: 3605 and 3510 (OH), 1749 and 1732 (split.) (CO), 1550 and 1345 (NO2). 1H-NMR (300 MHz, CDCl3) δ: 1.46 (3H, s, 3-CH3), 1.65 and 1.71 (each 3H, s, gem-CH3), 1.74 and 1.91 (each 1H, ddd, J=14, 9.5, 6 Hz, 4-H2), 1.99–2.22 (2H, m, 5-H2), 2.78 (1H, s, OH), 5.12 (1H, tsept, J=7, 1 Hz, 6-H), 5.34 and 5.41 (each 1H, d, J=17.5 Hz, 1-H2), 9.22 (2H, d, J=2 Hz, ArH), 9.26 (1H, t, J=2 Hz, ArH). 13C-NMR (75 MHz, CDCl3) δ: 17.73, 22.15, 25.68, 26.01, 39.69, 67.36, 79.53, 122.69, 123.04, 129.71 (C×2), 133.03, 133.49, 148.65 (C×2), 162.00, 200.26. HR-MS (ESI) m/z Calcd for C17H19O8N2 [M−H]− 379.1147. Found 379.1151.
(R)-12bIn the same manner as described above, compound 11b8) (1.00 g, 2.62 mmol) was converted into (R)-12b (949 mg, 95%) as a pale yellow viscous oil; [α]D24 +17.6 (c=1.02, CHCl3). HR-MS (ESI) m/z Calcd for C17H19O8N2 [M−H]− 379.1147. Found 379.1151.
(S)-1,3-Dihydroxy-3,7-dimethyloct-6-en-2-one (CPB Pheromone) (13a)To a solution of compound 12a (591 mg, 1.56 mmol) in MeOH (16 mL) was added Et2NH (85 µL, 0.78 mmol) at r.t. After being stirred at r.t. for 10 min, the mixture was concentrated. The resulting residue was purified by flash CC (AcOEt–hexane, 1 : 2) to provide compound 13a (274 mg, 95%) as a colorless oil. 1H- and 13C-NMR data were identical with those reported.22) The enantiomeric purity was confirmed (97% ee) by chiral HPLC [CHIRALPAK ID 0.46×25 cm (Daicel), EtOH–hexane (15 : 85), 1.0 mL/min, 30°C, 210 nm (detect.); 13a: 5.66 min, 13b: 6.73 min]; [α]D26 +3.6 (c=1.00, CHCl3). IR (CHCl3) cm−1: 3607 and 3610 (OH), 1712 (CO). 1H-NMR (300 MHz, CDCl3) δ: 1.37 (3H, s, 3-CH3), 1.58 and 1.67 (each 3H, s, gem-CH3), 1.71 and 1.80 (each 1H, ddd, J=14, 9.5, 5.5 Hz, 4-H2), 1.84–2.16 (2H, m, 5-H2), 2.97 (1H, s, OH), 2.97 (1H, t, J=5 Hz, OH), 4.46 and 4.53 (each 1H, d, J=20, 5 Hz, 1-H2), 5.04 (1H, t sept, J=7, 1 Hz, 6-H). 13C-NMR (75 MHz, CDCl3) δ: 17.66, 22.15, 25.63, 26.12, 39.92, 64.65, 78.46, 122.95, 133.31, 214.15. HR-MS (ESI) m/z Calcd for C10H18O3Na [M+Na]+ 209.1148. Found 209.1147.
(R)-13bIn the same manner as described above, compound 11b (683 mg, 1.80 mmol) was converted into (R)-13b (308 mg, 92%) as a colorless oil. The enantiomeric purity was confirmed (98% ee) by chiral HPLC: [α]D27 −2.4 (c=1.00, CHCl3). HR-MS (ESI) m/z Calcd for C10H18O3Na [M+Na]+ 209.1148. Found 209.1149.