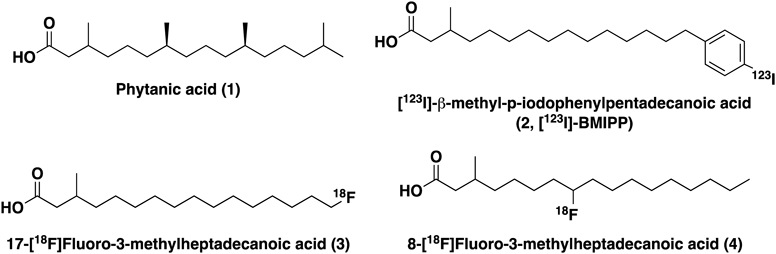
Experimental
General Methods and Materials1H- and 13C-NMR spectra were recorded on a JEOL ECA-500 instrument. Chemical shifts are reported in parts per million (ppm) relative to a tetramethylsilane internal standard (0.0 ppm) or to the solvent peak (CDCl3, δH 7.26 ppm, δC 77.1 ppm). 1H-NMR data are reported as follows: Chemical shift (δ ppm), integration, multiplicity, and coupling constant (J, Hz). 13C-NMR data are reported as follows: chemical shift (δ ppm), multiplicity, and coupling constant (Hz, where applicable). Multiplicities are reported using the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; sp, septet; m, multiplet; and br, broad. Only the strongest and/or structurally relevant IR peaks are reported (cm−1). All mass spectra were measured on a JEOL JMS-700 MStation mass spectrometer. Column chromatography was performed using silica gel (CHROMATOREX PSQ 100B, Fuji Silysia Chemical, Ltd.). Analytical thin layer chromatography was performed on glass plates pre-coated with silica gel (Merck, Kieselgel 60 F254, 0.25 mm).
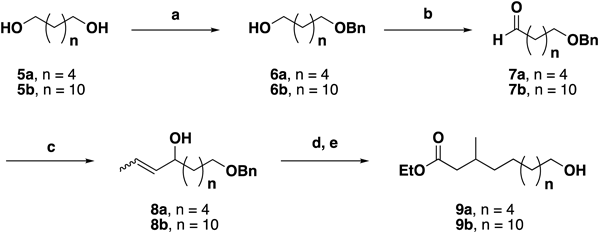
Synthetic Procedures6-(Benzyloxy)hexan-1-ol (6a)To a stirred solution of 1,6-hexanediol (5a, 5.91 g, 50.0 mmol) in tetrahydrofuran (THF, 200 mL) at 0°C was added NaH (2.00 g, 50.0 mmol). After 20 min, BnBr (5.20 mL, 43.8 mmol) was added, and the reaction mixture heated under reflux for 5 h. After this time, a saturated aqueous NH4Cl solution was added, and the reaction mixture was extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting crude product was purified by silica gel column chromatography (hexane–ethyl acetate, 3 : 1–1 : 1) to afford 6a as a clear colorless oil (4.96 g, 23.8 mmol, 54% yield based on BnBr). 6b (white wax, 8.94 g, 30.5 mmol, 44%) was synthesized from 1,12-dodecanediol (5b, 14.2 g, 70.0 mmol) according to the above procedure. The spectroscopic data obtained for 6a and b corresponded with literature values.11,12)
6-(Benzyloxy)hexanal (7a)To a stirred solution of 6a (4.96 g, 23.8 mmol) in CH2Cl2 (120 mL) at 0°C were added iodobenzene diacetate (11.5 g, 35.7 mmol) and TEMPO (743 mg, 4.76 mmol). After warming to 20°C and stirring for 15 h, saturated aq. Na2S2O3 was added, and the resulting mixture was extracted with CH2Cl2. The combined organic layer was then washed with saturated aq. NaHCO3 and brine, dried over anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting crude product was purified by silica gel column chromatography (hexane–ethyl acetate, 1 : 0–5 : 1) to afford 7a as a clear colorless oil (4.52 g, 21.9 mmol, 92%). 7b (white wax, 3.50 g, 12.1 mmol, 93%) was synthesized from 6b (3.80 g, 13.0 mmol) according to the above procedure. The spectroscopic data obtained for 7a and b corresponded with literature values.11,13)
(EZ)-9-(Benzyloxy)non-2-en-4-ol (8a)To a stirred solution of 7a (3.50 g, 17.0 mmol) in THF (85.0 mL) was added propenylmagnesium bromide (0.5 M in THF, 40.8 mL, 20.4 mmol) at −60°C. After stirring for 4 h, a 1 N HCl solution was added, and the reaction mixture was extracted with ethyl acetate. The combined organic layer was washed with 1 N HCl and brine, dried over anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting crude product was purified by silica gel column chromatography (hexane–ethyl acetate, 1 : 0–3 : 1) to afford 8a as a clear colorless oil (3.64 g, 14.7 mmol, 86%) and as an EZ mixture. 8b (pale yellow oil, 2.59 g, 7.79 mmol, 64%) was synthesized from 7b (3.50 g, 12.1 mmol) according to the above procedure. Data for 8a: 1H-NMR (CDCl3) δ: 1.27–1.69 (11H, m), 3.46 (2H, t, J=6.0 Hz), 4.01 (0.5H, q, J=6.5 Hz), 4.44 (0.5H, q, J=6.5 Hz), 4.49 (2H, s), 5.35–5.40 (0.5H, m), 5.43–5.49 (0.5H, m), 5.52–5.67 (1H, m), 7.25–7.35 (5H, m). 13C-NMR (CDCl3) δ: 13.26, 17.62, 25.10, 25.26, 26.08, 26.11, 29.63, 37.15, 37.31, 67.21, 70.29, 72.78, 72.98, 126.15, 126.69, 127.42, 127.56, 128.28, 133.52, 134.28, 138.57. IR (ATR) cm−1: 3403, 2935, 2858, 1453. FAB-MS m/z: 231.1774 (Calcd for C16H25O2–H2O: 231.1743). Data for 8b: 1H-NMR (CDCl3) δ: 1.27–1.70 (23H, m), 3.46 (2H, t, J=6.0 Hz), 4.02 (0.6H, q, J=6.5 Hz), 4.46 (0.4H, q, J=6.0 Hz), 4.50 (2H, s), 5.36–5.41 (0.4H, m), 5.44–5.50 (0.6H, m), 5.53–5.68 (1H, m), 7.27–7.35 (5H, m). 13C-NMR (CDCl3) δ: 13.30, 17.68, 25.29, 25.46, 26.14, 29.44, 29.54, 29.72, 37.24, 37.40, 67.33, 70.47, 72.79, 73.14, 126.15, 126.70, 127.42, 127.58, 128.30, 133.57, 134.34, 138.62. IR attenuated total reflectance (ATR) cm−1: 3364, 2923, 2852, 1454. FAB-MS m/z: 315.2712 (Calcd for C22H37O2–H2O: 315.2682).
Ethyl 10-Hydroxy-3-methyldecanoate (9a)To a stirred solution of 8a (3.64 g, 14.7 mmol) in triethyl orthoacetate (15.0 mL) was added pivalic acid (300 mg, 3.00 mmol) and the reaction mixture heated to reflux. After stirring for 19 h, the reaction mixture was concentrated in vacuo to give the crude target ethyl ester, which was used in the following stage without further purification. Thus, to a stirred solution of this crude mixture in ethanol (45.0 mL) was added Pd/C (10%, 2.40 g, 2.60 mmol). After stirring under an H2 atmosphere for 22 h at 20°C, the resulting mixture was filtered through a celite pad and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexane–ethyl acetate, 1 : 0–4 : 1) to afford 9a as a clear colorless oil (2.54 g, 11.0 mmol, 75% over 2 steps). 9b (clear colorless oil, 1.68 g, 5.34 mmol, 68%) was synthesized from 8b (2.59 g, 7.79 mmol) according to the above procedure. Data for 9a: 1H-NMR (CDCl3) δ: 0.93 (3H, d, J=6.0 Hz), 1.24–1.36 (13H, m), 1.57 (2H, quint. J=7.0 Hz), 1.89–1.99 (1H, m), 2.07–2.12 (1H, m), 2.26–2.31 (1H, m), 3.64 (2H, t, J=6.5 Hz), 4.13 (2H, q, J=7.0 Hz). 13C-NMR (CDCl3) δ: 14.23, 19.67, 25.65, 26.75, 29.32, 29.62, 30.29, 32.71, 36.61, 41.88, 60.06, 62.94, 173.40. IR (ATR) cm−1: 3456, 2855, 1734, 1463. FAB-MS m/z: 231.1989 (Calcd for C13H27O3: 231.1955). Data for 9b: 1H-NMR (CDCl3) δ: 0.93 (3H, d, J=6.0 Hz), 1.24–1.37 (25H, m), 1.57 (2H, quint. J=7.0 Hz), 1.89–1.99 (1H, m), 2.07–2.11 (1H, m), 2.27–2.31 (1H, m), 3.64 (2H, t, J=6.5 Hz), 4.13 (2H, q, J=7.0 Hz). 13C-NMR (CDCl3) δ: 14.26, 19.69, 25.71, 26.88, 29.40, 29.60, 29.72, 30.34, 32.78, 36.70, 41.93, 60.05, 63.05, 173.43. IR (ATR) cm−1: 3369, 2853, 1736, 1464. FAB-MS m/z: 315.2919 (Calcd for C19H39O3: 315.2894).
Ethyl 3-Methyl-10-oxodecanoate (10a)To a stirred solution of 9a (1.54 g, 6.70 mmol) in CH2Cl2 (33.0 mL) at 0°C were added iodobenzene diacetate (3.24 g, 10.0 mmol) and TEMPO (0.21 g, 1.34 mmol). After stirring for 8 h at this temperature, saturated aq. Na2S2O3 was added, and the reaction mixture extracted with CH2Cl2. The combined organic layer was washed with saturated aq. NaHCO3, dried over anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography (hexane–ethyl acetate, 1 : 0–10 : 1) to afford 10a as a clear colorless oil (981 mg, 4.30 mmol, 64%). 10b (clear colorless oil, 986 mg, 3.15 mmol, 93%) was synthesized from 9b (1.08 g, 3.40 mmol) according to the above procedure. Data for 10a: 0.93 (3H, d, J=6.5 Hz), 1.24–1.37 (m, 9H), 1.63 (2H, quint. J=7.0 Hz), 1.89–1.99 (1H, m), 2.07–2.12 (1H, m), 2.26–2.30 (1H, m), 2.43 (2H, td, J=7.0, 2.0 Hz), 4.13 (2H, q, J=7.0 Hz), 9.77 (1H, t, J=2.0 Hz). 13C-NMR (CDCl3) δ: 14.24, 19.65, 21.97, 26.63, 29.05, 29.34, 30.24, 36.53, 41.84, 43.84, 60.04, 173.29, 202.85. IR (ATR) cm−1: 2927, 2856, 2719, 1727, 1462. FAB-MS m/z: 229.1776 (Calcd for C13H25O3: 229.1798). Data for 10b: 1H-NMR (CDCl3) δ: 0.93 (3H, d, J=6.5 Hz), 1.24–1.35 (21H, m), 1.63 (2H, quint., J=7.0 Hz), 1.90–1.99 (1H, m), 2.07–2.11 (1H, m), 2.42 (2H, td, J=7.0, 1.5 Hz), 4.13 (2H, q, J=7.0 Hz), 9.77 (1H, t, J=1.5 Hz). 13C-NMR (CDCl3) δ: 11.94, 17.33, 19.68, 24.46, 26.75, 26.93, 27.00, 27.14, 27.18, 27.32, 27.91, 34.29. 39.53, 41.59, 57.69, 170.95, 200.66. IR (ATR) cm−1: 2924, 2853, 2711, 1730, 1464. FAB-MS m/z: 313.2738 (Calcd for C19H37O3: 313.2737).
Ethyl 10-Hydroxy-3-methylhexadecanoate (11a)To a stirred solution of 10a (400 mg, 1.80 mmol) in THF (9.00 mL) was added hexylmagnesium bromide (2.0 M in THF, 1.05 mL, 2.10 mmol) at −60°C. After warming to −40°C and stirring at this temperature for 2 h, 1 N HCl was added, and the reaction mixture was extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography (hexane–ethyl acetate, 1 : 0–15 : 1) to afford 11a as a white wax (403 mg, 1.28 mmol, 73%). 11b (white solid, 560 mg, 1.40 mmol, 47%) was synthesized from 10b (935 mg, 3.00 mmol) according to the above procedure. Both compounds 11a and b were obtained and characterized as diastereomeric mixtures. Data for 11a: 1H-NMR (CDCl3) δ: 0.89 (3H, t, J=7.5 Hz), 0.93 (3H, d, J=6.5 Hz), 1.24–1.46 (25H, m), 1.90–1.99 (1H, m), 2.07–2.13 (1H, m), 2.26–2.30 (1H, m), 3.57–3.59 (1H, m), 4.13 (2H, q, J=7.0 Hz). 13C-NMR (CDCl3) δ: 14.06, 14.25, 19.68, 22.59, 25.59, 26.80, 29.35, 29.61, 29.67, 30.31, 31.82, 36.64, 37.43, 37.46, 41.90, 60.05, 71.95, 173.39. IR (ATR) cm−1: 3322, 2925, 2854, 1735, 1460. FAB-MS m/z: 297.2781 (Calcd for C19H39O3–H2O: 297.2788). Data for 11b: 1H-NMR (CDCl3) δ: 0.89 (3H, t, J=7.5 Hz), 0.93 (3H, d, J=6.5 Hz), 1.24–1.46 (37H, m), 1.90–1.99 (1H, m), 2.07–2.11 (1H, m), 2.27–2.31 (1H, m), 3.54–3.60 (1H, m), 4.13 (2H, q, J=7.0 Hz). 13C-NMR (CDCl3) δ: 14.05, 14.24, 19.68, 22.58, 25.59, 25.63, 26.86, 29.35, 29.59, 29.68, 29.71, 30.32, 31.82, 36.69, 37.45, 41.90, 60.02, 71.94, 173.39. IR (ATR) cm−1: 3341, 2926, 2849, 1732, 1463. FAB-MS m/z: 381.3724 (Calcd for C25H51O3–H2O: 381.3727).