Review
Creation of Novel Cyclization Methods Using sp-Hybridized Carbon Units and Syntheses of Bioactive Compounds
2017 Volume 65 Issue 6 Pages 511-523
Details
2017 Volume 65 Issue 6 Pages 511-523
Some recent results on the development of new and reliable procedures for the construction of diverse ring systems based on the chemistry of sp-hybridized species, especially allene functionality, are described. This review includes: (i) synthesis of the multi-cyclic skeletons by combination of the π-component of allene with suitable other π-components such as alkyne, alkene, or additional allene under Rh-catalyzed conditions; (ii) synthesis of heterocycles as well as carbocycles by reaction of the sp-hybridized center of allene with some nucleophiles in an endo-mode manner; and (iii) total syntheses of natural products and related compounds from the sp-hybridized starting materials.
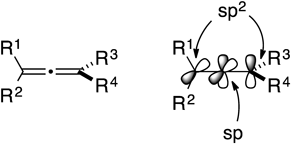
We have been investigating the chemistry of alkynes, a typical (sp-sp)-hybridized two carbon species, particularly focusing on the Nicholas reaction and Pauson–Khand reaction of alkyne derivatives for over two decades. Those accomplishments were summarized in several previous reviews.1–7) On the other hand, another representative sp-hybridized substrate, the allenes (Fig. 1), have an accumulated diene structure consisting of the unique (sp2-sp-sp2)-hybridized three-carbon unit, which not only creates a significantly high reactivity but also provides a wide variety of possible reaction modes. We envisioned that appropriate and selective use of one (distal or proximal) of the two π-components of the allene functionality would open an alternative door for new cyclization methods. Thus we started out the project regarding the development of the new ring-closing reaction by taking advantage of the inherent properties of allenes. In this review, we summarize some successful results that have been accumulated in our laboratory during the last decade. In addition, total syntheses of bioactive compounds and their related ones using the sp-hybridized starting materials are described.
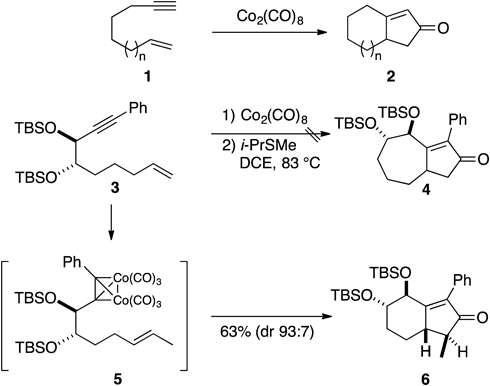
The Pauson–Khand reaction is a carbonylative [2+2+1] cycloaddition of alkyne, alkene, and carbon monoxide (CO) resulting in the formation of cyclopentenone derivatives. The intramolecular version of this reaction is well known as one of the most reliable and straightforward methods for the construction of bicyclo[3.3.0]octenones 2 (n=0) and bicyclo[4.3.0]nonenones 2 (n=1) from the corresponding ene-ynes 1. However, it was found that this reaction is not applicable to the efficient preparation of larger-sized bicyclic species such as bicyclo[5.3.0]decenones 2 (n=2). We encountered difficulties in synthesizing the bicyclo[5.3.0] derivative 4 from the ene-yne 3.8) Indeed, when the Co2(CO)6-complexed alkyne 5, derived from the reaction of 3 with Co2(CO)8, was exposed to isopropyl methyl sulfide, none of target compound 4 could be detected in the reaction mixture, and the unexpected bicyclo[4.3.0]nonenone derivative 6 was produced in a highly stereoselective manner (Chart 1).
We envisioned that the allene functionality having an accumulated diene structure would become an alternative π-component of alkene moiety in the Pauson–Khand-type [2+2+1] carbonylative cycloaddition so as to overcome the insufficiency encountered in preparation of the bicyclo[5.3.0]decenone 4. During our investigations on the carbonylative [2+2+1] cycloaddition of allenes, we mainly used the 3-phenylsulfonyl-substituted allene-ynes as substrates due to their ready availability as well as their ability to selectively react at the distal π-bond. Thus 8-trimethylsilyl-3-phenylsulfonyl-octa-1,2-diene-7-yne (7) was treated with a catalytic amount of [RhCl(CO)dppp]2 (dppp: 1,3-bis(diphenylphosphino)propane) in refluxing toluene under CO atmosphere to afford the bicyclo[4.3.0]nonadienone 8 in 97% yield, which must have been formed by exclusively taking part in the distal double bond of allene.9–11) The other possible isomer, the proximal product 9, could not be detected. This RhI-catalyzed [2+2+1] carbonylative cycloaddtion of allene-yne 7 could be expanded to various kinds of one-carbon homologated substrates 10 using [RhCl(CO)dppp]2 and/or [RhCl(CO)2]2 leading to the efficient construction of bicyclo[5.3.0]decadienone derivatives 11. This RhI-catalyzed carbonylative reaction9–11) requires neither a geminal dialkyl substituent effect nor a template effect. The ester functionality as well as hydroxyl and siloxy groups can be tolerated,9–11) and both azabicyclic and oxabicyclic frameworks12,13) are constructed as expected. Some examples9–13) are shown in Chart 2.

Our endeavors then turned to the application of this RhI-catalyzed [2+2+1] carbonylative cycloaddition to the construction of one-carbon enlarged bicyclo[6.3.0] skeletons.14) The simple linear allene-yne 12 (Z=H) and allene-yne 12 (Z=CO2Me) in refluxing xylene underwent the ring-closing reaction to give the bicyclo[6.3.0] derivatives 13 in 23 and 43% yield, respectively. Higher yields were obtained when allene-ynes 14 (R=H and Ph) were exposed to RhI-catalyst to produce the benzene-fused bicyclo[6.3.0]undecadienones 15 (R=H: 90% and R=Ph: 83% yield) (Chart 3). Formation of the one-carbon shortened bicyclo[4.3.0]nonadienones was easily accomplished by this method as expected.14)

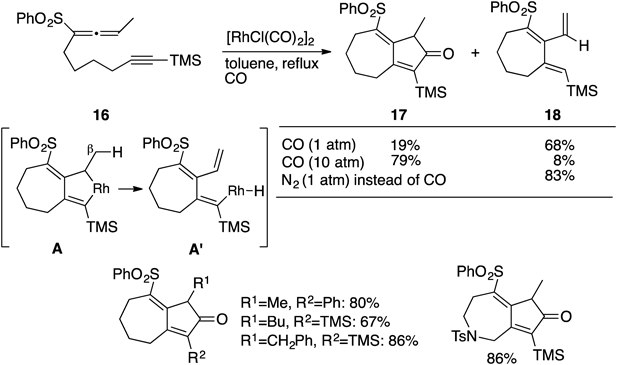
When the 1,3,3-trisubstitued allene derivative 16 was exposed to the standard conditions, the desired bicyclo[5.3.0] derivative 17 was obtained in 19% yield. The seven-membered triene 18 was found to be the major product (68%).12,13) This drawback could immediately be solved by increase of pressure of CO. Indeed, on treatment under 10 atm of CO, 16 produced the [2+2+1] cycloaddition product 17 in 79% yield along with the triene 18 in 8% yield. The formation of triene 18 could be rationalized in terms of the intermediacy of the common rhodabicyclic species A, which would undergo β-hydride elimination to furnish the triene 18 through reductive elimination of the intermediate A′. The other successful examples12,13) are depicted in Chart 4. As can be expected by consideration of the reaction mechanism, the triene 18 was exclusively formed in 83% yield when the reaction was carried out under N2 atmosphere instead of CO. On the other hand, it should be mentioned that the bicyclo[4.2.0] frameworks 20 were easily synthesized by heating the allene-ynes 19 in xylene in the absence of RhI-catalyst under N2 atmosphere. Both bicyclo[5.2.0] and bicyclo[6.2.0] derivatives15) could also be produced by simple heating in xylene or mesitylene; some of these are shown in Chart 5. The formation of these bicyclo[m.2.0] compounds might be rationalized by the biradical intermediates B.

The carbonylative [2+2+1] cycloaddition has been limited almost exclusively to the use of alkyne as one of π-components. The promising results from the carbonylative [2+2+1] cycloaddition of allene-ynes prompted us to investigate RhI-catalyzed carbonylative [2+2+1] cycloaddition of allene-enes.16) Based on the established conditions for allene-ynes (Charts 2 and 3), allene-ene 21 was treated with RhI-catalyst under 5 atm of CO in toluene at 120°C to afford the bicyclo[4.3.0]nonenone derivative 22 in 96% yield as a mixture of trans- and cis-isomers in a ratio 79 : 21 via isomerization of the initially formed 22′ by participation of the distal double bond of the allene. Alternatively, modified conditions (addition of AgBF4 under a low CO pressure)17) also worked well, in particular, when the substrates have 1,1-disustituted alkene as exemplified by conversion of 23 into 24. Allene-enes having an electron-withdrawing group on the allenyl moiety such as 21 tend to produce cyclopentenone derivatives, whereas alkyl-substituted ones such as 23 afford cyclopentanone frameworks. This RhI-catalyzed [2+2+1] carbonylative cycloaddition of allene-enes could be applied to the construction of not only the seven-membered bicyclo[5.3.0]decenone but also the more complex tricyclic derivative (Chart 6).

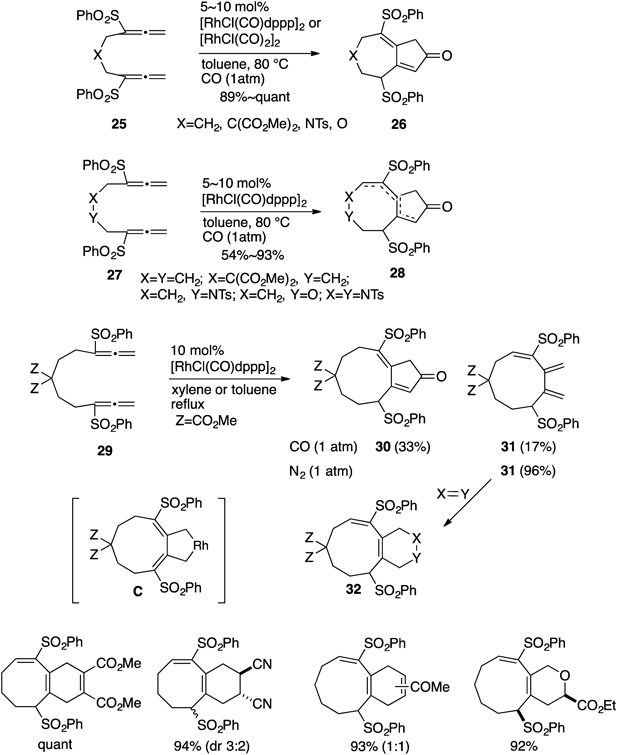
It was clearly shown that changing one of the two π-components (alkyne or alkene) of ene-ynes to an allene group led to significant improvement of yields, particularly in the case of the formation of bicyclo[5.3.0] frameworks. These results would be considered mainly due to use of the allene functionality. However, efficient formation of the corresponding eight-membered structures from allene-ynes (and -enes) was not consistently attained except for the ones possessing template effects. We envisioned that larger-sized bicyclo[6.3.0]undecadienones might be synthesized in satisfactory yields if the two π-components of bisallene species could participate in the ring-closing process. Thus our next efforts were directed toward RhI-catalyzed [2+2+1] carbonylative cycloaddition of bisallenes.18–20) The initial attempt was performed by treatment of bisallenes 25 with a catalytic amount of [RhCl(CO)2]2 or [RhCl(CO)dppp]2 to afford the bicyclo[5.3.0]decadienones 26 in high yields. The larger-sized eight-membered bicyclo[6.3.0]undecadienones 28 were obtained from 27 in satisfactory-to-high yields.18–20) A more complicated tricyclic (6/8/5) compound was prepared by this method.21) Furthermore, it would become interesting to see whether construction of the nine-membered bicyclo[7.3.0]dodecadienone skeletons 3020) would be possible using this method. The bisallene 29 was exposed to RhI-catalyst in refluxing xylene to give the desired 30 in a rather low yield (33%) along with the nine-membered monocyclic compound 31 possessing a bis(exo-methylene) group (17%). Exclusive formation of 31 was realized when 29 was exposed to RhI-catalyzed conditions under N2 atmosphere instead of CO to furnish 31 in 96% yield.20) The common intermediate C would be considered for the production of both 30 and 31 (Chart 7). The thus formed monocyclic compound 31 subsequently underwent [4+2]-type cycloaddition with suitable dienophiles22) resulting in the formation of bicyclo[m.4.0] derivatives 32 in high yields (Chart 7).
The [2+2+1] carbonylative cycloaddition of allenes developed in our laboratory can be summarized as follows. RhI-catalyzed reaction of allene-ynes consistently produced the bicyclo[5.3.0]decadienones (seven-membered compounds) in satisfactory-to-high yields, whereas the classical Pauson–Khand reaction of ene-ynes generally afforded the bicyclo[5.3.0] frameworks in unsatisfactory yields. We also showed that allene-enes produced the bicyclo[5.3.0] frameworks in good yields. However, allene-ynes and allene-enes were not suitable substrates for preparation of bicyclo[6.3.0]undecadienones (eight-membered compounds) except for specific cases where substrates have a template effect such as a benzene ring. This limitation can be completely overcome by introduction of an additional allene group to the substrates. Thus bicyclo[6.3.0]undecadienones could be obtained in high yields when bisallenes were exposed to RhI-catalyzed standard conditions. It should be emphasized that even nine-membered bicyclo[7.3.0]dodecadienone was also prepared, although the yield was unsatisfactory.
1.4. Hetero-[2+2+1] Carbonylative CycloadditionThe carbodiimide group is N(sp2)-C(sp)-N(sp2) functionality and regarded as an isoelectronic alternative to the allenyl moiety in the [2+2+1] carbonylative cycloaddition of allene-ynes. After screening several catalytic conditions,23,24) we found that the pyrrolo[2,3-b]indol-2-one skeleton 34 could be prepared when carbodiimide-yne derivatives 33 were treated with a catalytic amount of Co2(CO)8 (30 mol%) and tetramethylthiourea (TMTU) (30 mol%) in toluene. Several successful examples are depicted in Chart 8. Thus it became clear that the carbodiimide functionality could serve as an alternative to the allene group in [2+2+1] carbonylative cycloaddition. On the other hand, the nitrile group, which is formally regarded as an aza-congener of an alkyne group, is frequently used as the reactive aza-π-component in the [2+2+2] cycloaddition reaction leading to pyridine derivatives. In sharp contrast to those results, the nitrile group cannot be used in the [2+2+1] carbonylative cycloaddition as π-component. We expected that a combination of nitrile with allene might work well to yield the corresponding five-membered lactams. Thus treatment of allene-nitriles 3525) with 10 mol% [RhCl(CO)dppp]2 in toluene under CO atmosphere afforded benzo[f]oxyindoles 36. This reaction could be applied to the non-benzene ring-containing aliphatic compounds 37 to produce the azabicyclo[3.3.0] derivatives 3825) (Chart 8). The larger-sized azabicyclo[m.3.0] derivatives (m=4, 5) could be constructed by this method, although their yields were low. We tentatively understood that these reactions would proceed via the in situ formed allene-ketenimine species 39 on the basis of several experiments (Chart 8).

Benzene-1,2-bisallene and ene-bisallene species could be prepared from the corresponding bis(propargyl alcohol)s and expected to undergo 6π-electrocyclic reaction leading to formation of 2,3-naphthoquinodimethane and o-quinodimethane intermediates. On the basis of this prediction, we investigated the following reactions.
2.1. Allene–Benzene–AllenesOn treatment with dialkyl(chloro)phosphanes in tetrahydrofuran (THF) under mild conditions (from −78°C to room temperature), the benzene-bridged bis(propargyl alcohol)s 4026,27) underwent sequential dual formation of (propargyl ester) and its [2,3]-sigmatropic rearrangement to form the bisallene 41, which immediately collapsed to the 2,3-naphthocyclobutene derivative 42 via the predicted 6π-electrocyclic reaction and cyclobutene ring formation. Several examples are shown in Chart 9.

Based on the results in 2.1,26,27) we envisioned that efficient generation of o-quinodimethanes 44 could be realized if ene-bis(propargyl alcohol) derivatives 43 were employed as starting materials. The 1,3-diene moiety of the in situ-formed o-quinodimethanes 44 would be intramolecularly or intermolecularly captured by suitable dienophiles to afford 45. Indeed, the ene-bis(propargyl alcohol) derivative 46, for example, was treated with phenylsulfenyl chloride in the presence of dimethyl fumarate to provide the tetralin derivative 47 in 73% overall yield (including treatment with meta-chloroperoxybenzoic acid [m-CPBA] to simplify structure determination).28–30) The o-quinodimethane intermediate (e.g., 44) reacted with a variety of dienophiles irrespective of the properties of the olefinic counterpart,28–30) although it was obvious that electron-deficient olefins showed much higher reactivity than electron-rich ones. The intramolecular version of the above [4+2] cycloaddition of the ene-bis(propargyl alcohol) derivatives efficiently afforded tricyclic compounds as exemplified by transformation of 48 into 49 (Chart 10). It is noteworthy that this tandem procedure was applicable to the synthesis of steroid skeleton 50.28–30)

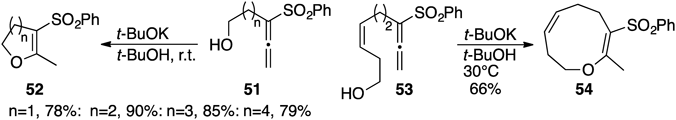
The phenylsulfonylallenes 51 (n=1) having a hydroxyethyl group underwent endo-mode ring-closing reaction31–33) to produce exclusively the corresponding dihydrofurans 52 (n=1) in 78% yield when treated with t-BuOK. The six- to eight-membered oxacycles 52 (n=2,3,4)31–34) could also be prepared in high yields under standard conditions. Introduction of (Z)-double bond on the alkyl tether of 53 enabled to construct even the nine-membered tetrahydrooxonin 54 in 66% yield.34) Diethyl phosphonate functionality could be employed instead of phenylsulfonyl group34) as an electron-withdrawing group in this reaction (Chart 11).
A similar endo-mode ring-closing reaction occurred when the terminal nucleophile of 51 was changed from hydroxyl group to nitrogen ones. In the case of the azide derivatives 55,35) the five- and six-membered azacyclic compounds 56 and 57 were obtained under reductive conditions; however, the seven-membered one could not be formed. On the other hand, on treatment with base, the amide derivatives 58 provided six- and seven-membered enamides 5935) (Chart 12).

This endo-mode ring-closing reaction was applicable to carbon analogues.36,37) Indeed, the substrates 60 having an active methine moiety at the ω-position of alkyl tether produced the five- to seven-membered carbocycles 61. Production of 61 could be tentatively interpreted by the intermediacy of the initially formed carbocycles 62, which would easily release one of two electron-withdrawing groups. This plausible mechanism was in part supported by several additional experiments.37) Several examples are shown in Chart 13.

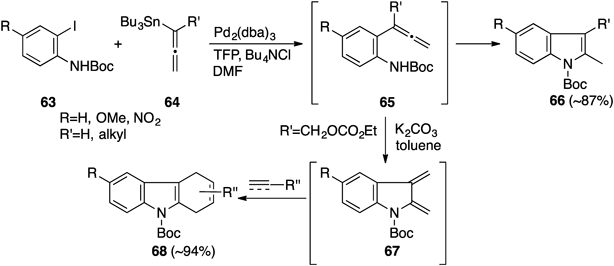
A new procedure for the construction of the 2,3-disubstituted-indole nucleus from allenylaniline derivatives was developed based on allene chemistry. Treatment of 2-iodoanilines 63 with allenylstannanes 64 (R′=alkyl) under Stille coupling conditions (Pd2(dba)3 [dba: dibenzylideneacetone], tri-2-furylphosphine [TFP], and tetrabutylammonium chloride) directly afforded 2,3-disubstituted indoles 66.38–40) The formation of 66 could be rationalized by initial Pd-catalyzed coupling reaction, followed by endo-mode ring-closing reaction of the resulting allenylaniline derivatives 65. Indeed, 65 could be isolated under milder conditions and transformed into 66.38–40) We postulated that introduction of a suitable leaving group into R′ of the allenylaniline derivatives 65 would accelerate SN2′-type displacement by amino functionality at the sp-hybridized center leading to in situ formation of indole-2,3-quinodimethane such as 67, which would be subsequently captured by suitable dienophiles. Thus allenylaniline 65 (R=H, R′=CH2OCO2Et),41,42) which was prepared by the Stille coupling reaction between 63 and 64, was treated with a catalytic amount of K2CO3 in toluene to give the carbazole derivatives 68. We thereby developed a new method for generation of indole-2,3-quinodimethane (Chart 14).
The terminal hydrogen of allene group is acidic when a suitable electron-withdrawing group is in residence at the 3-position. We envisioned that intramolecular trapping of allenyl anions generated by base treatment of the substrates 69, prepared from 51 in Chart 11, might produce the corresponding cyclic compounds 70 having alkyne appendage. On the basis of our simple analysis,43,44) we examined the ring-closing reaction of 69 as depicted in Chart 15. The iodo or tosylate derivatives 69 were exposed to tetrabutylammonium fluoride (TBAF) or sodium hydride (NaH) in N,N-dimethylformamide (DMF) at 0°C to furnish the three- to seven-membered carbocycles 70 in high yields. Predictably, the electron-withdrawing group on the allenyl moiety was essential in this transformation and no reaction occurred when a phenylsulfonyl group on this moiety was exchanged to alkyl groups.43,44)

Highly strained cyclopropane derivatives have served as useful and powerful C3-building blocks for the construction of various ring systems. In sharp contrast to the extensive investigation of the RhI-catalyzed [5+2] cycloaddition of vinylcyclopropanes with π-counterparts, very few examples of the ring-closing reaction of allenylcyclopropanes have been reported. We investigated the intramolecular cycloaddition of allenylcyclopropane–alkyne derivatives 71.45) Treatment of 71 with RhI-catalyst effected not only the ring opening of cyclopropane but also cycloisomerization to produce bicyclo[5.4.0]undecatrienes 72. In some cases, small amounts of the cyclopentenylidene derivatives 73 were isolated. The formation of both 72 and 73 could be rationalized by the common rhodacyclohexenylidene intermediate 74, which should arise by aid of the release of the high strain energy (27.5 kcal/mol) of cyclopropane. The intermediate 74 would in turn collapse to 72 via successive insertion of alkyne group into the C–Rh bond and reductive elimination. Alternatively, direct reductive elimination of 74 must result in the production of 73. The seven/seven-membered bicyclo[5.5.0]dodecatrienes 75 could also be prepared by this ring-closing reaction45) (Chart 16).

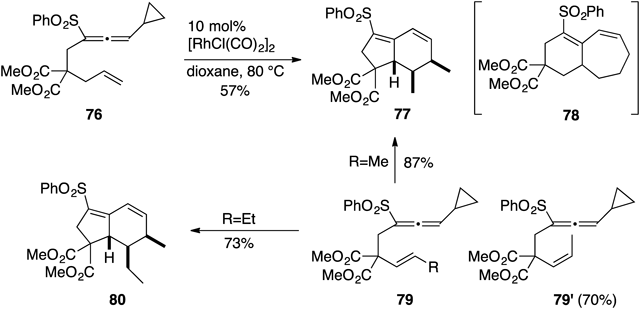
The easy conversion of 71 into 72 made us imagine easy preparation of the dihydro derivatives of 72 using an alkene counterpart instead of alkyne group. Therefore we next investigated cycloaddition of allenylcyclopropanes with alkenes.46) Allenylcyclopropane–alkene 76 was exposed to 10 mol% of [RhCl(CO)2]2 in dioxane at 80°C to produce bicyclo[4.3.0]nonadiene 77 in 57% yield in an exclusively stereoselective manner. The bicyclo[5.4.0] skeleton 78, being expected on the basis of conversion of 71 into 72, could not be detected. We postulated that isomerization of an allyl substituent to a propenyl group would occur before (or after) forming the rhodacyclohexenylidene intermediate (e.g., 74 in Chart 16). To demonstrate our hypothesis, the internal alkene 79 (R=Me) was exposed to standard conditions to afford 77 in higher yield (87%). The corresponding (Z)-isomer 79′ also furnished 77 in 70% yield. Those two experiments using propenyl derivatives obviously disclosed that this transformation is highly stereoselective, but not strereospecific. The ethyl congener 79 (R=Et) gave 80 in 73% yield. In order to understand the mechanism for production of 77 from 76, 79, and 79′, we performed several experiments using deuterated substrates and found that this transformation involves domino-type reaction and should provide new insights into allene chemistry46) (Chart 17).
The observation that exchange of the reactive alkyne part of 71 by an alkene group (i.e., 76 or 79) drastically changed the reaction pathway prompted us to investigate RhI-catalyzed cycloaddition of allenylcyclopropane–allenes.47) On exposure to 10 mol% [RhCl(CO)dppp]2 in toluene at 80°C, the allenylcyclopropane–allenes 81 were transformed into the bicyclo[5.3.0]decatrienes 82 accompanied by evolution of ethylene. The cyclopentenylidene derivatives 83 were obtained as a major product in most cases. Although the chemical yields of the 1,5,6,7-tetrahydroazulene 82 are far from satisfactory, this transformation is unique and again quite different from those of allenylcyclpopropane–alkyne and –alkene derivatives. Thus cyclopropyl group on the allenyl moiety served as C1-unit in this ring-closing reaction, which might be regarded as a new formal [5+2−2]-type cycloisomerization47) (Chart 18).

The results obtained in the cycloaddition of allenylcyclopropanes under RhI-catalyzed conditions can be simply summarized as follows. The allenylcyclopropane reacted with an additional π-component in the presence of RhI-catalyst to provide three types of products depending on the property of an additional π-counterpart. Namely, allenylcyclopropane–alkynes produced the bicyclo[5.4.0]undecatrienes via the [5+2] cycloaddition process. The bicyclo[4.3.0]nonadiene skeletons became the major products when allenylcyclopropane–alkenes were exposed to RhI-catalyzed conditions. The reaction pathway of this transformation is more complicated, but highly stereoselective and non-stereopsecific. In the case of allenylcyclopropane–allenes, the formal [5+2–2]-type cycloisomerization occurred with liberation of ethylene.
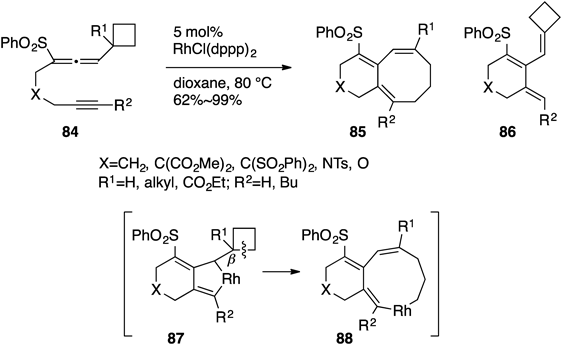
4.4. [6+2] Cycloaddition of Allenylcyclobutane–AlkynesFunctionalized cyclobutanes such as cyclobutanones and hydroxy- and alkylidenecyclobutanes are widely used as acyclic C4-building blocks, whereas the simplest cyclobutane without any other functional groups on its ring is rarely employed. The easy cleavage of the unfunctionalized cyclopropane ring that was activated by the adjacent allenyl moiety (described in 4.1) prompted us to examine RhI-catalyzed intramolecular [6+2] cycloaddition reaction between alkyne and the one-carbon homologated allenylcyclobutane leading to the formation of bicyclo[6.4.0] frameworks.48) Conversion of allenylcycobutane–alkyne 84 into bicyclo[6.4.0]dodecatrienes 85 could be realized by treatment with 5 mol% of RhCl(dppp)2 in dioxane at 80°C. The formation of 85 was tentatively interpreted by the pathway via the rhodabicyclo[4.3.0]nonadiene intermediate 87. The cyclobutane ring of 87 is opened by a β-carbon elimination, which releases the ring strain energy (26.3 kcal/mol) to generate the nine-membered intermediate 88, the reductive elimination of which would result in the formation of 85. When the starting material 84 possesses no substituent at the allenic position (R1=H), β-hydride elimination proceeds to produce 86 as a minor pathway. In this ring-closing reaction, functionalized cyclobutanes were not mandatory, but the simplest cyclobutane could serve as a suitable four-carbon unit48) (Chart 19).
Since the strain energies of the normal-sized cycloalkanes, namely cyclopentane and cyclohexane, are considered too low (6.3 and approximately 0 kcal/mol, respectively), use of these rings as acyclic C5- or C6-building blocks has not been reported. Encouraged by the successful development of the Csp3–Csp3 bond activation of the simplest unactivated cyclobutane described in 4.4, we next investigated the application of this methodology to the analogous cyclopentane derivatives.49) After screening various conditions, we found that treatment of allenylcyclopentane–alkynes 89 with Wilkinson catalyst produced the bicyclo[7.4.0] compound 90 via the unprecedented Csp3–Csp3 bond cleavage of the unactivated cyclopentane. The formation of 90 is tentatively rationalized in terms of the intermediacy of the rhodabicyclo[4.3.0]nonadiene intermediate 92, which would be derived from the RhI-catalyzed cycloaddition between allene and alkyne moieties of 89. The intermediate 92 might subsequently collapse to the bicyclo[7.4.0]tridecatriene derivatives 90 via β-carbon elimination (Csp3–Csp3 bond activation) in good yields. Changing the RhI-catalyst to RhCl(dppp)2 brought the different reaction mode to produce the spiro[2.4]heptane derivatives 91. The formation of the spiro[2.4]heptane derivatives 91 could be explained by Cγ–H elimination (Csp3–H bond activation) of the common intermediate 9249) (Chart 20).

In 4.4 and 4.5 we described how the unprecedented Csp3–Csp3 bond activation of less-reactive cycloalkanes (simple cyclobutanes and cyclopentanes) could be achieved by RhI-catalyzed cycloadditions of 84 and 89. On the basis of the plausible mechanisms proposed for these reactions, we thought that the allene–alkyne unit works as a highly reactive π-component toward the RhI-catalyst to form the common rhodabicyclic intermediate, which can activate a C–C and/or C–H bond near the Rh center. Next, we succeeded in developing another type of C–H activation of benzylallene–alkynes using this concept.50) Treatment of 93 with RhI-catalyst effected novel cycloisomerization to produce tricyclo[9.4.0.03,8]pentadecapentaene derivatives 94 through Csp2–H bond activation. Based on deuteration and competition experiments, a reaction mechanism was proposed, which might involve a vinylidenecarbene–Rh intermediate 9550) (Chart 21).

Transition metal-catalyzed [2+2+2] cycloaddition of allene components could be a useful methodology for constructing complex skeletons; however, this has been less fully explored than the analogous reactions of alkynes or alkenes. Consideration of the rhodabicyclic intermediate of Section 4 led us to envisage that the allene–alkyne derivatives 96 possessing an additional π component would react with RhI-catalyst to form the intermediates 98. Insertion of the remaining π-component into the C–Rh bond followed by reductive elimination would then afford the three-membered ring products 97.51) Treatment of 96 with [RhCl(CO)2]2 effected the intramolecular [2+2+2]-type ring-closing reaction to produce various tetracyclic derivatives 97 containing a cyclopropane ring. The substrates with acyclic alkenes could also be used resulting in the formation of the tricyclic derivatives. Experiments using the trans-alkene 99 and cis-alkene 100 derivatives clearly indicated that this intramolecular [2+2+2] cycloaddition is highly stereoselective as well as stereospecific51) (Chart 22).

We successfully accomplished total syntheses of more than twenty natural products and their related compounds by taking advantage of the inherent properties of the sp-hybridized substrates. These structures are shown in Chart 23.

This review briefly summarized our recent results on the development of new and reliable procedures for construction of diverse ring systems based on the chemistry of sp-hybridized species, especially allene functionality. Combination of the π-component of allene with other suitable π-components such as alkyne, alkene, or additional allene under Rh-catalyzed conditions enabled us to build up multi-cyclic skeletons that might, in general, be harder to obtain by standard procedures. On the other hand, the sp-hybridized center of allene tends to be attacked by some nucleophiles in an endo-mode manner resulting in the formation of heterocycles as well as carbocycles. Eventually, total syntheses of natural products and their related compounds were completed using the sp-hybridized starting materials.
This review of the author’s work was written by the author upon receiving the 2016 Pharmaceutical Society of Japan Award.
The achievements reported in this review were due to the consistently enthusiastic efforts of the talented and devoted former and present co-workers whose names are cited in the references. This work was supported by not only KAKENHI from JSPS but also a Grant-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology of Japan, for which we are greatly thankful.
The author declares no conflict of interest.