Current Topics : Reviews
Cyclic ADP-Carbocyclic-Ribose and -4-Thioribose, as Stable Mimics of Cyclic ADP-Ribose, a Ca2+-Mobilizing Second Messenger
2018 Volume 66 Issue 2 Pages 155-161
Details
2018 Volume 66 Issue 2 Pages 155-161
Cyclic ADP-ribose (cADPR), a general mediator involved in Ca2+ signaling, has the characteristic 18-membered ring consisting of an adenine, two riboses and a pyrophosphate, in which the two primary hydroxy groups of the riboses are linked by a pyrophosphate unit. This review focuses on chemical synthetic studies of cADPR analogues of biological importance. Although cADPR analogues can be synthesized by enzymatic and chemo-enzymatic methods using ADP-ribosyl cyclase, the analogues obtained by these methods are limited due to the substrate-specificity of the enzymes. Consequently, chemical synthetic methods providing a greater variety of cADPR analogues are required. Although early chemical synthetic studies demonstrated that construction of the large 18-membered ring structure is difficult, the construction was achieved using the phenylthiophosphate-type substrates by treating with AgNO3 or I2. This is now a general method for synthesizing these types of biologically important cyclic nucleotides. Using this method as the key step, the chemically and biologically stable cADPR mimic, cADP-carbocyclic-ribose (cADPcR) and -4-thioribose (cADPtR), were synthesized.
Cyclic ADP-ribose (cADPR, 1, Fig. 1), a metabolite of nicotinamide adenine dinucleotide (NAD+), was originally isolated from sea urchin by Lee and colleagues.1) Thereafter, it has been demonstrated that cADPR mobilizes intracellular Ca2+ in various mammalian cells, such as, pancreatic β-cells, smooth muscle, cardiac muscle, T-lymphocytes, and cerebellar neurons, and therefore cADPR is now recognized as a general mediator of intracellular Ca2+ signaling.2–8)

Lee et al. also confirmed the structure of cADPR by X-ray crystallographic analysis, which is the characteristic 18-membered cyclic structure including a pyrophosphate linkage.9) Although pyrophosphate linkages, e.g., the linkage in NAD+, are often readily cleaved enzymatically,10) cADPR is resistant to pyrophosphatases from various sources,11) probably due to its unusual cyclic structure. However, treatment of cADPR with t-BuOK in dimethyl sulfoxide (DMSO) selectively cleaved the pyrophosphate linkage.11) Under neutral conditions, cADPR is in a zwitterionic form with a positive charge around the N1-C6-N6 moiety (pKa=8.3), making the molecule extremely unstable (Chart 1). The charged adenine moiety attached to the anomeric carbon of the ribose can be an efficient leaving group so that cADPR is readily hydrolyzed at the unstable N1-ribosyl linkage of its adenine moiety to produce ADP-ribose (ADPR, 4), even in neutral aqueous solution.10) Under physiological conditions, cADPR is also hydrolyzed at the N1-ribosyl linkage by cADPR hydrolase to give the inactive ADPR.10)

A number of cADPR analogues have been designed and synthesized, since they are potentially useful for investigating the mechanism of cADPR-mediated Ca2+ release.12–14) As described above, cADPR is biologically as well as chemically unstable, which has limited studies of its physiological role, at least to some extent. Therefore stable equivalents of cADPR exhibiting a Ca2+-mobilizing activity similar to that of cADPR are very useful as tools in biological studies as well as potential drug lead structures.
In this minireview, our studies on the design, synthesis, and biological activity of cADPR analogues to develop its stable equivalents resulting in identification of cyclic ADP-carbocyclic-ribose (cADPcR, 2) and cyclic ADP-4-thioribose (cADPtR, 3) are summarized.
cADPR analogues have been synthesized predominantly by enzymatic and chemo-enzymatic methods using ADP-ribosyl cyclase-catalyzed cyclization15–22) (Chart 2). A significant drawback of the enzymatic and chemo-enzymatic procedures is that these methods require preparation of the nicotinamide dinucleotide-type precursors as the substrates and the synthesis of these precursors is often troublesome. More importantly, the analogues that can be obtained by these methods are limited due to the substrate-specificity of the ADP-ribosyl cyclase.
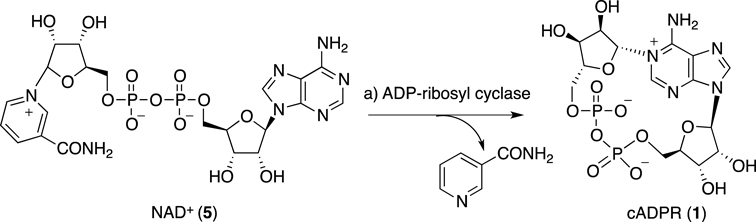
The total synthesis of cADPR itself may not be of importance, since cADPR is easily prepared by enzymatic synthesis from β-NAD+(Chart 2). On the other hand, because of the limitations of the enzymatic and chemo-enzymatic methods described above, effective chemical synthetic methods for the preparation of a greater variety of cADPR analogues are of importance.
cADPR (1) has the large 18-membered ring consisting of an adenine, two β-riboses attaching to the adenine N1 and N9 positions, and a pyrophosphate that connects the two primary hydroxyl groups of the riboses. In the synthesis of cADPR and its analogues, construction of the characteristic 18-membered ring structure should be the key step. The highly polar phosphate moiety seems to be detrimental to purification and reactions, and therefore it should be introduced at a later stage in the synthetic process. Early studies on the chemical synthesis of cADPR and its analogues had been extensively performed to cralyfy that construction of the characteristic pyrophosphate-containing 18-membered ring structure is rather difficult.23–26)
We designed cyclic ADP-carbocyclic-ribose (cADPcR, 2) and its inosine congener cyclic inosine 5′-diphosphate (IDP) carbocyclic-ribose (cIDPcR, 6, Fig. 2) as stable mimics of cADPR, in which the ring oxygen in the N1-ribose is replaced by a methylene to lack the unstable N1-ribosyl linkage of cADPR. Thus we expected that these carbocyclic analogues should be resistant to both enzymatic and chemical hydrolysis, while still preserving most of the functional groups of cADPR, except for the ring oxygen.

We first tried to construct the 18-membered ring structure in the synthesis of inosine congener cIDPcR (6), because construction of the N1-substituted hypoxanthine structure in cIDPcR seemed to be easier than that of the corresponding charged N1-substituted adenine structure in cADPcR. The construction of the N1-carbocyclic-ribosylinosine structure was achieved by treating N1-(2,4-dinitrophenyl)inosine derivative 7, prepared by Piccialli’s method,27) with the chiral carbocyclic amine 8,24) followed by silylation of the primary hydroxy group and subsequent basic treatment for the adenine ring-closure reaction (Chart 3).

Hata and Sekine reported that intermolecular condensation between a phosphorothioate and a phosphomonoester was effectively promoted by I2 or AgNO3 to give the corresponding pyrophosphate product.28,29) We expected that intramolecular version of their method might effectively provide the desired 18-membered pyrophosphate ring. Thus, after conversion of 9 into phosphorothioate 12, we found that when 12 was treated with I2 and MS3A in pyridine, the desired cyclization product 13 was obtained in excellent yield.30,31) Acidic treatment of 13 to remove the isopropylidene groups finally provided cIDPcR (6).
cIDPcR showed only an insignificant Ca2+-mobilizing effect in sea urchin egg homogenates, however, we successfully developed a very efficient method for synthesizing cIDPcR (6), which became an entry to a general method for synthesizing cADPR analogues, as described below.
Compare with cIPDcR (6) lacking the amino function of the adenine, cADPcR (2) preserves all of the functions of cADPR except for replacing the N1-ribose ring oxygen with a methylene, so that we expected it to act as a cellular Ca2+-mobilizing agent like cADPR.
With the above encouraging results in mind, we next attempted the total synthesis of cADPcR (2). Development of an efficient method for constructing the N1-carbocyclic-ribosyladenosine structure was key for completing the synthesis of cADPcR. It turned out that treatment of a mixture of imidazole nucleoside 15, readily prepared from the known imidazole nucleoside 14, and chiral carbocyclic-ribosyl amine 8 with K2CO3 in MeOH gave the desired N1-carbocyclic-ribosyladenine product 16 in high yield32) (Chart 4). Compound 16 was converted into the corresponding phosphorothioate-type substrate 17, which was treated with AgNO3/Et3N/MS 3A in pyridine to give the desired cyclization product 18 in 93% yield. Acidic deprotection of 16 finally furnished cADPcR (2).32)

cADPcR was not only both chemically and biologically stable, but also effectively mobilizes intracellular Ca2+ in sea urchin eggs32) and is more potent than cADPR in neuronal cells.33) In addition, cADPcR induced insulin secretion from digitonin-permeabilized rat islets via Ca2+-mobilization like as cADPR. However, to our surprise, it was inactive in T cells, whereas natural cADPR effectively mobilizes Ca2+ in both neuronal cells and T cells.34) These results confirmed that the target proteins and/or the mechanism of action of cADPR in sea urchin eggs, T cells, and neuronal cells are different,35) as suggested by previous biological studies.36–42)
The abovementioned results on cADPcR brought up a question that “why is cADPcR active in neuronal cells and sea urchin eggs like cADPR but inactive in T cells unlike cADPR?”
cADPR is in equilibrium between the N6-protonated amino-form and the N6-deprotonated imino-form, as shown in Chart 1.43) One difference between cADPR and cADPcR is their pKa values for protonation at the N6-position. The pKa of cADPcR (8.9)32) is somewhat higher than that of cADPR (8.3),43) which might affect its interaction with target proteins. Under physiological conditions, cADPR can be in a mixture of the protonated amino form and the deprotonated imino form, where cADPcR should be present mostly in the amino-form due to its relatively higher pKa. This pKa difference between cADPR and cADPcR might be related to their activities in T-cells.
In cADPR and its analogues, the most stable conformation can be the one with minimal steric repulsion between the adenine moiety and both of the N1- and N9-ribose moieties. It should be noted that, in cADPcR, the C6″-Hβ, which is absent in cADPR, is very sterically repulsive to the adenine C2-H (Fig. 3). Accordingly, the stable conformation of cADPcR is likely to differ from that of cADPR due to the steric effect, which might also affect its interaction with the target proteins.

Based on these results and considerations, we designed cADPtR (3, Fig. 1), a 4-thioribose analogue of cADPR.44) It has been recognized that 4′-thionucleosides can be useful bioisosteres of the natural nucleosides, in which the 4-thioribose does not only effectively mimic the ribose of nucleosides,45–51) but also the N-4-thioriobosyl linkage is more stable than the corresponding N-ribosyl linkage against both the chemical and enzymatic hydrolysis.52,53) In addition, the pKa value of cADPtR is expected to be similar to that of cADPR due to the electron-withdrawing property of sulfur atom. Furthermore, the conformation of cADPtR, particularly spatial positioning of the N1-thioribose and adenine moieties, would be similar to that of cADPR, because both the sulfur and oxygen have a similar sp3 configuration bearing two non-bonding electron pairs. Thus, based on the pKa and steric features, we expected that cADPtR might be a stable cADPR equivalent that is active in various cells including T cells as is cADPR.
The restrosynthetic analysis for cADPtR (3) is shown in Fig. 4. In the synthesis of cADPtR, stereoselective construction of the unknown N1-β-thioribosyladenosine structure was the key issue. We speculated that if the 4-thioribosylamine derivative 19 would be in equilibrium between the α-anomer 19α and the β-anomer 19β, stereoselective construction of the N1-β-thioribosyladenosine structure might be achieved, because the relatively less reactive α-anomer 19α would not undergo the condensation, but rather would be effectively converted into the more reactive β-anomer 19β via the equilibrium to result in accumulation of the desired β-condensation product 20β (Chart 5). Indeed, treatment of 19 (α/β-mixture), which was prepared from a known 1-deoxythioribose 2154) (Chart 6), with imidazole nucleoside 15 (2.1 eq) in MeOH at room temperature produced the desired β-product 20β in 61% yield concomitant with 5% of the α-product 20α, as we expected.



Next, the construction of the 18-membered cyclic pyrophosphate structure was achieved, using the phosphorothioate 25 as a substrate converted from 20β, by the Ag+-promoted intramolecular condensation, similarly to that in the synthesis of cADPcR to afford the cyclization product 26. Thus we successfully synthesized the target cADPtR (3), after acidic deprotection of 26 (Chart 7).

The pKa of cADPtR was determined to be 8.0, which was similar to that of cADPR (pKa=8.3)43) and about 1 unit lower than that of cADPcR (pKa=8.9).32) Structural analysis of cADPR (1), cADPcR (2), and cADPtR (3) by molecular dynamics calculations using a simulated annealing method based on the nuclear Overhauser effect (NOE) constraints clarified the structural differences in detail. The three obtained structures were superimposed, revealing that the cADPtR structure (red) resembles that of cADPR (blue) but the cADPcR structure (green) is not similar to those of the other two compounds, as shown in Fig. 5. Particularly, the relative special arrangement of the N1-carbocyclic ribose and the adenine of cADPcR clearly differs from those of the other two compounds. We confirmed that the cADPR structure solved by X-ray crystallographic analysis resembles the calculated cADPR structures, which suggests our computational structure determination was appropriate (Fig. 5b). Therefore, the pKa and conformational properties of cADPtR precisely mimic those of cADPR.
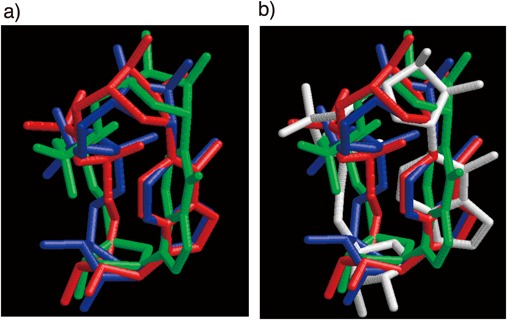
Only heavy atoms are shown. a) Three calculated structures are superimposed: cADPR (blue), cADPcR (green), and cADPtR (red). b) Four structures are superimposed: The crystal structure of cADPR (white), the calculated structures of cADPR (blue), cADPcR (green), and cADPtR (red).
cADPtR is, unlike cADPR, chemically and biologically stable, while it effectively mobilizes intracellular Ca2+ similar to cADPR in various biological systems, such as sea urchin homogenate, NG108-15 neuronal cells, and Jurkat T-lymphocytes. Thus cADPtR is a stable equivalent of cADPR,44) which can be useful as a biological tool for investigating cADPR-mediated Ca2+-mobilizing pathways.
We have successfully developed an efficient method for the chemical synthesis of cADPR analogues, in which the construction of the 18-membered pyrophosphate-ring structure using the Ag+-promoted intramolecular condensation reaction with an S-phenyl phosphorothioate-type substrate is the key step. Using the method, we successfully identified cADPcR and cADPtR as stable analogues of cADPR, which can be useful as an effective tool for identifying target proteins and for investigating cADPR-mediated signaling pathways. The chemical synthetic method is now generally employed for synthesizing these types of biologically important cyclic nucleotides particularly those chemically modified in the N-1-linked ribose moiety, which are not expected to be accessible by an enzymatic route.55–66)
The author is grateful to Ms. Michiyo Shirato, Mr. Yuji Sumita, Dr. Masayoshi Fukuoka, Dr. Takashi Kudoh, Ms. Natsumi Sakaguchi, Mr. Masayoshi Tsuzuki, Mr. Takatoshi Sato, Mr. Satoshi Takano, Prof. Yoshihito Ueno and Prof. Akira Matsuda for their extensive study and discussion as co-workers at Hokkaido University, and to Prof. Haruhiro Higashida (Kanazawa University), Prof. Takashi Murayama (Juntendo University), Prof. Barry B. L. Potter (University of Bath), Prof. Andrea Guse (Humber University), and Dr. Tomoshi Kameda (AIST, Japan) for their helpful collaborations.
The author declares no conflict of interest.