Experimental
Chemistry1H-NMR spectra were recorded on a Varian 400-MR and BRUKER AV-III HD500, and chemical shifts were expressed as δ (ppm) values with tetramethylsilane as an internal reference (s=singlet, d=doublet, t=triplet, m=multiplet, dd=double doublet, dt=double triplet, ddd=double double doublet, and br=broad peak). MS were recorded on a Waters ultra performance liquid chromatography (UPLC)/SQD and Waters Acquity UPLC/ZQ. Electrospray ionization (ESI) positive high-resolution (HR)-MS were obtained using a Thermo EXACTIVE-Plus Waters LCT Premier.
2-Methyl-5-[2-(quinolin-2-yl)ethyl]pyrazolo[1,5-a]pyrimidin-7-ol (3)A mixture of ethyl potassium malonate (1019 mg, 6.00 mmol), magnesium chloride (571 mg, 6.00 mmol) and triethylamine (1070 µL, 7.68 mmol) in tetrahydrofuran (THF) (24 mL) was stirred at room temperature for 2.5 h. One milliliter of the reaction mixture was added to another reaction mixture of 3-(quinoline-2-yl)propanoic acid (17.3 mg, 0.100 mmol) and 1,1′-carbonyldiimidazole (CDI; 24.3 mg, 0.150 mmol) in THF (0.2 mL) which had been stirred at 50°C for 2 h. The combined reaction mixture was stirred at 50°C for 15 h. After cooling at room temperature, to the reaction mixture were added 1 M HCl aqueous solution (0.5 mL), CHCl3 and saturated NaHCO3 aqueous solution (0.5 mL). The mixture was through the phase separator. The organic layer was concentrated in vacuo. To the residue was added 3-methyl-1H-pyrazole-5-amine (9.70 mg, 0.100 mmol) in 1,4-dioxane (0.100 mL) and AcOH (5.0 µL, 87 µmol), and the mixture was stirred at 90°C for overnight. After cooling at room temperature, the mixture was concentrated in vacuo. The residue was purified by preparative HPLC (Waters SunFire™ Column, C18, 5 µm, 19×100 mm, 10–95% MeOH in 0.1% (v/v) formic acid aqueous solution) to give 3 (5.4 mg, 17%) as a pale brown solid. 1H-NMR (DMSO-d6) δ: 2.27 (s, 3H), 3.09 (dd, 2H, J=9.0, 6.8 Hz), 3.32–3.36 (m, 2H), 5.57 (d, 1H, J=1.4 Hz), 5.95 (s, 1H), 7.49 (d, 1H, J=8.4 Hz), 7.56 (ddd, 1H, J=8.0, 6.9, 1.2 Hz), 7.74 (ddd, 1H, J=8.4, 6.9, 1.5 Hz), 7.93–7.96 (m, 2H), 8.30 (d, 1H, J=8.3 Hz), 12.23 (s, 1H); MS (ESI) m/z 305 [M+H]+; HR-MS (ES+) Calcd for C18H17ON4 [M+H]+ 305.1397; Found, 305.1398.
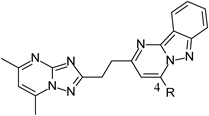
2-Methyl-5-(quinolin-2-yl)pyrazolo[1,5-a]pyrimidin-7-ol (4)Compound 4 was prepared from quinaldic acid in a manner similar to that described for compound 3, with a yield of 12%. 1H-NMR (DMSO-d6) δ: 2.34 (s, 3H), 6.22 (s, 1H), 6.72 (s, 1H), 7.75 (ddd, 1H, J=8.1, 7.0, 0.99 Hz), 7.92 (ddd, 1H, J=8.4, 6.9, 1.4 Hz), 8.12 (d, 1H, J=7.5 Hz), 8.24 (d, 1H, J=8.4 Hz), 8.37 (d, 1H, J=8.8 Hz), 8.62 (d, 1H, J=8.8 Hz), 12.53 (s, 1H); MS (ESI) m/z 277 [M+H]+; HR-MS (ES+) Calcd for C16H13ON4 [M+H]+ 277.1084; Found, 277.1084.
2-Methyl-5-[(quinolin-2-ylsulfanyl)methyl]pyrazolo[1,5-a]pyrimidin-7-ol (5)Compound 5 was prepared from (quinolin-2-ylsulfanyl)acetic acid in a manner similar to that described for compound 3, with a yield of 27%. 1H-NMR (DMSO-d6) δ: 2.26 (s, 3H), 4.52 (s, 2H), 5.84 (s, 1H), 6.00 (s, 1H), 7.47 (d, 1H, J=8.6 Hz), 7.54 (ddd, 1H, J=8.0, 7.0, 1.1 Hz), 7.76 (ddd, 1H, J=8.4, 7.0, 1.4 Hz), 7.90–7.98 (m, 2H), 8.23 (d, 1H, J=8.6 Hz), 12.43 (br s, 1H); MS (ESI) m/z 323 [M+H]+; HR-MS (ES+) Calcd for C17H15ON4S [M+H]+ 323.0961; Found, 323.0961.
2-Methyl-5-[2-(pyridin-2-yl)ethyl]pyrazolo[1,5-a]pyrimidin-7-ol (6)A mixture of ethyl potassium malonate (2145 mg, 12.6 mmol), magnesium chloride (1428 mg, 15.0 mmol) and triethylamine (2676 µL, 19.2 mmol) in THF (60 mL) was stirred at room temperature for 2.5 h. Half milliliter of the reaction mixture was added to another reaction mixture of 3-(pyridin-2-yl)propanoic acid (7.56 mg, 50.0 µmol) and CDI (12.2 mg, 75 µmol) in THF (0.2 mL) which had been stirred at 50°C for 2 h. The combined reaction mixture was stirred at 50°C for overnight. After cooling at room temperature, to the reaction mixture were added 1 M HCl aqueous solution (0.5 mL), CHCl3 (2.5 mL) and saturated NaHCO3 aqueous solution (0.5 mL). The mixture was through the phase separator. The organic layer was concentrated in vacuo. To the residue was added 3-methyl-1H-pyrazole-5-amine (4.90 mg, 50 µmol) in 1,4-dioxane (0.100 mL) and AcOH (5.0 µL, 87 µmol), and the mixture was stirred at 90°C for overnight. After cooling at room temperature, the mixture was concentrated in vacuo. The residue was purified by preparative HPLC (Waters SunFire™ Column, C18, 5 µm, 19×100 mm, 10–95% MeOH in 0.1% (v/v) formic acid aqueous solution) to give 6 (5.3 mg, 42%). MS (ESI) m/z 255 [M+H]+.
2-Methyl-5-[2-([1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]pyrazolo[1,5-a]pyrimidin-7-ol (7)Compound 7 was prepared from 3-([1,2,4]triazolo[1,5-a]pyrimidine-2-yl)propanoic acid in a manner similar to that described for compound 3, with a yield of 40% as a pale brown solid. 1H-NMR (DMSO-d6) δ: 2.27 (s, 3H), 3.05–3.13 (m, 2H), 3.23–3.30 (m, 2H), 5.57 (s, 1H), 5.94 (s, 1H), 7.31 (dd, 1H, J=6.8, 4.3 Hz), 8.83 (dd, 2H, J=4.3, 1.9 Hz), 9.33 (dd, 1H, J=6.8, 1.9 Hz), 12.20 (s, 1H); MS (ESI) m/z 296 [M+H]+; HR-MS (ES+) Calcd for C14H14ON7 [M+H]+ 296.1254; Found, 296.1257.
5-[2-(Imidazo[1,2-a]pyridin-2-yl)ethyl]-2-methylpyrazolo[1,5-a]pyrimidin-7-ol (8)Compound 8 was prepared from 3-(imidazo[1,2-a]pyridin-2-yl)propanoic acid in a manner similar to that described for compound 6, with a yield of 30%. MS (ESI) m/z 294 [M+H]+.
5-[2-(Imidazo[1,2-a]pyrimidin-2-yl)ethyl]-2-methylpyrazolo[1,5-a]pyrimidin-7-ol (9)Compound 9 was prepared from 3-(imidazo[1,2-a]pyrimidin-2-yl)propanoic acid in a manner similar to that described for compound 6, with a yield of 10%. MS (ESI) m/z 295 [M+H]+.
5-[2-(1H-Benzimidazol-2-yl)ethyl]-2-methylpyrazolo[1,5-a]pyrimidin-7-ol (10)Compound 10 was prepared from 3-(1H-benzimidazole-2-yl)propanoic acid in a manner similar to that described for compound 3, with a yield of 27%. 1H-NMR (DMSO-d6) δ: 2.27 (s, 3H), 3.09 (t, 2H, J=7.5 Hz), 3.23 (t, 2H, J=7.3 Hz), 5.54 (s, 1H), 5.95 (s, 1H), 7.08–7.16 (m, 2H), 7.43–7.53 (m, 2H), 12.30 (br s, 2H); MS (ESI) m/z 294 [M+H]+; HR-MS (ES+) Calcd for C16H16ON5 [M+H]+ 294.1349; Found, 249.1347.
3-(5,7-Dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)propanoic Acid (23)A mixture of aminoguanidine hydrochloride 21 (25.0 g, 226 mmol) and succinic anhydride 22 (25.0 g, 250 mmol) was stirred at 190°C for 1 h. After cooling at room temperature, to the reaction mixture were added sodium hydroxide (27.0 g, 675 mmol) and H2O (67 mL). The mixture was stirred at 120°C for 1.5 h. After cooling, to the reaction mixture were added conc. HCl aqueous solution (37 mL) and H2O (25 mL) at room temperature, and the mixture was stood overnight. The resulting precipitate under ice-bath cooling was collected by filtration. To the residue were added EtOH (400 mL), acetylacetone (20.0 mL, 194 mmol) and AcOH (2.00 mL, 35.0 mmol), and the mixture was stirred under reflux for 7 h. After cooling at room temperature, to the mixture were added H2O (30 mL) and sodium hydroxide (10.0 g, 250 mmol), and the mixture was stirred at room temperature for 2 h. The mixture was filtered and the filtrate was concentrated in vacuo lightly. To the solution was added conc. HCl aqueous solution (20 mL) and the mixture was cooled under ice-bath. The resulting precipitate was collected by filtration to give 23 (33.7 g, 68%) as a colorless solid. 1H-NMR (DMSO-d6) δ: 2.55 (s, 3H), 2.69 (d, 3H, J=0.9 Hz), 2.76 (t, 2H, J=7.3 Hz), 3.05 (t, 2H, J=7.4 Hz), 7.10 (d, 1H, J=0.9 Hz), 12.20 (br s, 1H); MS (ESI) m/z 221 [M+H]+.
5-[2-(5,7-Dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]-2-methylpyrazolo[1,5-a]pyrimidin-7-ol (11)To a mixture of 23 (661 mg, 3.00 mmol) in THF (10 mL) was added CDI (584 mg, 3.60 mmol), and the mixture was stirred at 50°C for 1 h. To the reaction mixture were added ethyl potassium malonate (1.02 g, 6.00 mmol), magnesium chloride (571 mg, 6.00 mmol), triethylamine (1.04 mL, 7.50 mmol), and the mixture was stirred at 50°C for 12 h. After cooling at room temperature, to the reaction mixture was added 1 M HCl aqueous solution, and the mixture was stirred at ambient temperature for 1 h. The mixture was extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. To the residue were added 1,4-dioxane (10 mL), 5-methyl-1H-pyrazol-3-amine (200 mg, 2.06 mmol) and AcOH (1.18 mL, 20.6 mmol), and the mixture was stirred at 90°C for 12 h. After cooling at room temperature, the mixture was diluted with Et2O, and the resulting precipitate was collected by filtration to give 11 (614 mg, 92%) as a beige solid. 1H-NMR (DMSO-d6) δ: 2.27 (s, 3H), 2.56 (s, 3H), 2.68 (d, 3H, J=1.0 Hz), 3.01–3.11 (m, 2H), 3.20–3.28 (m, 2H), 5.57 (s, 1H), 5.94 (s, 1H), 7.12 (d, 1H, J=0.8 Hz), 12.20 (br-s, 1H); MS (ESI) m/z 324 [M+H]+; HR-MS (ES+) Calcd for C16H18ON7 [M+H]+ 324.1567; Found, 324.1569.
2-Methyl-5-[2-(6-methyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]pyrazolo[1,5-a]pyrimidin-7-ol (12)To a mixture of 3-(6-methyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)propanoic acid (619 mg, 3.00 mmol) in THF (10 mL) was added CDI (584 mg, 3.60 mmol), and the mixture was stirred at 50°C for 1 h. To the reaction mixture were added ethyl potassium malonate (1.02 g, 6.00 mmol), magnesium chloride (571 mg, 6.00 mmol), triethylamine (1.04 mL, 7.50 mmol), and the mixture was stirred at 50°C for 12 h. After cooling at room temperature, to the reaction mixture was added 1 M HCl aqueous solution, and the mixture was stirred at ambient temperature for 1 h. The mixture was extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. To the residue were added 1,4-dioxane (10 mL), 5-methyl-1H-pyrazol-3-amine (200 mg, 2.06 mmol) and AcOH (1.18 mL, 20.6 mmol), and stirred at 90°C for 12 h. After cooling at room temperature, the mixture was diluted with Et2O, and the resulting precipitate was collected by filtration to give 12 (487 mg, 76%) as a beige solid. 1H-NMR (DMSO-d6) δ: 2.26 (s, 3H), 2.36 (d, 3H, J=0.8 Hz), 3.01–3.11 (m, 2H), 3.20–3.29 (m, 2H), 5.55 (s, 1H), 5.93 (s, 1H), 8.73 (d, 1H, J=2.4 Hz), 9.17 (dd, 1H, J=2.3, 1.1 Hz), 12.20 (s, 1H); MS (ESI) m/z 310 [M+H]+; HR-MS (ES+) Calcd for C15H16ON7 [M+H]+ 310.1411; Found, 310.1414.
2-[2-(5,7-Dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]pyrimido[1,2-b]indazol-4-ol (13)To a mixture of 23 (2.20 g, 10.0 mmol) in THF (33 mL) was added CDI (1.95 g, 12.0 mmol), and the mixture was stirred at 50°C for 1 h. To the reaction mixture were added ethyl potassium malonate (3.40 g, 20.0 mmol), magnesium chloride (1.90 g, 20.0 mmol), triethylamine (3.48 mL, 25.0 mmol), and the mixture was stirred at 50°C for 12 h. After cooling at room temperature, to the reaction mixture was added 1 M HCl aqueous solution (50 mL), and the mixture was stirred at ambient temperature for 1 h. The mixture was extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. To a half of the residue were added 1,4-dioxane (15 mL), 1H-indazol-3-amine (610 mg, 4.58 mmol) and AcOH (4 mL), and stirred at 95°C for 18 h. After cooling at room temperature, the mixture was concentrated in vacuo. The residue was diluted with EtOAc and saturated NaHCO3 aqueous solution, and the resulting precipitate was collected by filtration to give 13 (194 mg, 11%) as a beige solid. 1H-NMR (DMSO-d6) δ: 2.56 (s, 3H), 2.70 (d, 3H, J=0.7 Hz), 3.03–3.13 (m, 2H), 3.21–3.27 (m, 2H), 5.76 (s, 1H), 6.82 (ddd, 1H, J=7.9, 6.8, 0.8 Hz), 7.09 (d, 1H, J=0.9 Hz), 7.24–7.33 (m, 1H), 7.44 (d, 1H, J=8.6 Hz), 7.90 (dt, 1H, J=8.2, 1.0 Hz); MS (ESI) m/z 360 [M+H]+; HR-MS (ES+) Calcd for C19H18ON7 [M+H]+ 360.1567; Found, 360.1568.
4-Chloro-2-[2-(5,7-dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]pyrimido[1,2-b]indazole (17)A mixture of 13 (280 mg, 0.779 mmol) and phosphorous oxychloride (3.35 g, 21.8 mmol) was stirred at 100°C for 1 d. After cooling at room temperature, the mixture was concentrated in vacuo. The residue was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was washed with brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 0–3% MeOH in CHCl3) to give 17 (112 mg, 38%) as a yellow solid. 1H-NMR (DMSO-d6) δ: 2.55 (s, 3H), 2.68 (d, 3H, J=0.9 Hz), 3.36–3.58 (m, 4H), 7.10 (d, 1H, J=0.9 Hz), 7.32 (ddd, 1H, J=8.3, 6.7, 0.7 Hz), 7.67 (ddd, 1H, J=8.7, 6.7, 1.1 Hz), 7.87 (dt, 1H, J=8.6, 0.9 Hz), 7.98 (s, 1H), 8.23 (dt, 1H, J=8.3, 1.0 Hz); MS (ESI) m/z 378 [M+H]+; HR-MS (ES+) Calcd for C19H17N7Cl [M+H]+ 378.1228; Found, 378.1230.
2-[2-(5,7-Dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]-N-methylpyrimido[1,2-b]indazol-4-amine (14)To a solution of 17 (96.0 mg, 0.254 mmol) in THF (0.96 mL) was added methylamine solution (381 µL, 0.762 mmol) and the mixture was stirred at room temperature for 17 h. The mixture was concentrated in vacuo, and the residue was purified by flash column chromatography (silica gel, 0–20% MeOH in CHCl3) to give 14 (92.0 mg, 97%) as a yellow solid. 1H-NMR (DMSO-d6) δ: 2.56 (s, 3H), 2.70 (d, 3H, J=0.7 Hz), 3.04 (d, 3H, J=4.9 Hz), 3.33–3.43 (m, 4H), 6.58 (s, 1H), 7.05–7.14 (m, 2H), 7.43–7.58 (m, 1H), 7.66 (d, 1H, J=8.6 Hz), 8.05–8.13 (m, 1H), 8.14–8.23 (m, 1H); MS (ESI) m/z 373 [M+H]+; HR-MS (ES+) Calcd for C20H21N8 [M+H]+ 373.1884; Found, 373.1886.
2-[2-(5,7-Dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]-4-methoxypyrimido[1,2-b]indazole (15)To a solution of 17 (100 mg, 0.265 mmol) in MeOH (1.00 mL) was added sodium methoxide (28.6 mg, 0.529 mmol) and the mixture was stirred at room temperature for 3 h. The reaction mixture was diluted with H2O, and the resulting precipitate was collected by filtration. The residue was purified by flash column chromatography (silica gel, 0–10% MeOH in CHCl3). The residue was washed with EtOAc to give 15 (81.0 mg, 82%) as a yellow solid. 1H-NMR (DMSO-d6) δ: 2.56 (s, 3H), 2.70 (d, 3H, J=0.9 Hz), 3.39–3.55 (m, 4H), 4.28 (s, 3H), 7.11 (d, 1H, J=0.9 Hz), 7.14–7.29 (m, 2H), 7.56 (ddd, 1H, J=8.8, 6.7, 1.1 Hz), 7.72 (dt, 1H, J=8.7, 0.9 Hz), 8.15 (dt, 1H, J=8.3, 1.0 Hz); MS (ESI) m/z 374 [M+H]+; HR-MS (ES+) Calcd for C20H20ON7 [M+H]+ 374.1724; Found, 374.1726.
2-[2-(5,7-Dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]-4-methylpyrimido[1,2-b]indazole (16)To a stirred mixture of 17 (50.0 mg, 0.132 mmol), trimethylboroxine (49.8 mg, 0.397 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride dichloromethane adduct (Pd(dppf)Cl2·CH2Cl2; 32.4 mg, 39.7 µmol) in 1,4-dioxane (1.00 mL) was added K2CO3 (110 mg, 0.794 mmol) under argon atmosphere, and the mixture was stirred at 90°C for 1 d. After cooling at room temperature, the mixture was diluted with water and extracted with CHCl3. The organic layer was washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 0–5% MeOH in CHCl3) to give 16 (16.0 mg, 34%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 2.55 (s, 3H), 2.69 (d, 3H, J=0.7 Hz), 2.88 (d, 3H, J=0.7 Hz) 3.36–3.54 (m, 4H), 7.10 (d, 1H, J=0.9 Hz), 7.24 (ddd, 1H, J=8.2, 6.7, 0.8 Hz), 7.43–7.65 (m, 2H), 7.70–7.90 (m, 1H), 8.20 (dt, 1H, J=8.3, 0.9 Hz); MS (ESI) m/z 358 [M+H]+; HR-MS (ES+) Calcd for C20H20N7 [M+H]+ 358.1775; Found, 358.1776.
2-[2-(5,7-Dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)ethyl]pyrimido[1,2-b]indazole-4-carbonitrile (18)A mixture of 17 (50.0 mg, 0.132 mmol), potassium cyanide (18.0 mg, 0.276 mmol) and 1,4-diazabicyclo[2.2.2]octane (DABCO; 18.0 mg, 0.160 mmol) in 1,4-dioxane (2.0 mL) and H2O (0.20 mL) was stirred at room temperature for 5 h. The residue was diluted with H2O. The resulting precipitate was collected by filtration and purified by flash column chromatography (silica gel, 0–5% MeOH in CHCl3) to give 18 (18.0 mg, 37%) as a bright red solid. 1H-NMR (DMSO-d6) δ: 2.55 (s, 3H), 2.68 (s, 3H), 3.40–3.60 (m, 4H), 7.10 (s, 1H), 7.36–7.50 (m, 1H), 7.73 (ddd, 1H, J=8.4, 7.0, 1.0 Hz), 7.94 (d, 1H, J=8.6 Hz), 8.26–8.36 (m, 1H), 8.40 (s, 1H); MS (ESI) m/z 369 [M+H]+; HR-MS (ES+) Calcd for C20H17N8 [M+H]+ 369.1571; Found, 369.1571.
PDE10A Enzyme Assay Protocol
Expression and Purification of PDE10A Protein (for Inhibition Assay of Compounds 2–10)Human PDE10A phosphodiesterase domain (amino acids 449–789) was cloned between the NdeI and XhoI sites of pET28a vector to express target protein as a 6×histidine (His)-tagged protein. This plasmid was transformed in Escherichia coli BL21(DE3) and overexpressed. The cells were disrupted by sonication and the supernatant was collected by centrifugation at 4°C. The target protein was precipitated from the supernatant by adding ammonium sulfate to 60% and was collected by centrifugation. The precipitate was dissolved with nickel–nitrilotriacetic acid (Ni–NTA) wash buffer (25 mM Tris–HCl pH 8.0, 500 mM NaCl, 20 mM imidazole) and was applied onto an Ni–NTA column. After washing with Ni–NTA wash buffer, the bound protein was eluted with 25 mM Tris–HCl pH 8.0, 500 mM NaCl, 250 mM imidazole. Eluate was collected and the buffer exchanged with 25 mM Tris–HCl pH 8.0, 500 mM NaCl. The amino-terminal His tag of PDE10A (449–789) was removed by incubation with thrombin protease for 12 h at 4°C. After digestion, protein solution was passed through Benzamidine Sepharose 6B column (GE Healthcare, U.S.A.) and again applied onto an Ni–NTA column to remove thrombin and uncleaved His-tag protein. Further purification was performed by passing through Q Sepharose column (GE Healthcare) equilibrated with 25 mM Tris–HCl pH 8.0, 150 mM NaCl followed by Superdex 75 pg 16/60 (GE Healthcare) in 25 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES)-Na pH 7.5, 100 mM NaCl.
PDE10A Inhibition Assay (for Inhibition Assay of Compounds 2–10)Inhibition of human PDE10A enzyme activity was assessed by measuring the quantity of cAMP via the Homogeneous Time-Resolved Fluorescence (HTRF) detection method. The assay was performed in 20 µL samples containing an optimal amount of the PDE10A enzyme domain, a buffer (50 mM Tris–HCl pH 8.0; 2 mM MgCl2, 0.1% BSA), 0.1 µM cAMP and various concentrations of compounds (0.03 nM to 40 µM). After compounds were mixed with the enzyme, the reaction was initiated by adding the substrate cAMP and the mixture was incubated for 30 min at room temperature with agitation. The reaction was terminated on addition of the fluorescence acceptor (cAMP labeled with the dye d2) and the fluorescence donor (anti-cAMP antibody labeled with Cryptate, Cisbio). After 60 min, the fluorescence transfer corresponding to the amount of residual cAMP was measured at lex. 320 nm, lem. 620 nm and lem. 665 nm using an Envision plate reader (PerkinElmer, Inc., U.S.A.) and signal ratio (665/620) was calculated. The ratio determined in the absence of enzyme was subtracted from all data. The obtained results were converted to inhibition percentage relative to an uninhibited control.
PDE10A Inhibition Assay (for Inhibition Assay of Compounds 11–18)Inhibitory activity of compounds on human PDE10A2 was assessed by measuring the residual amount of cAMP, substrate for PDE10A2, by the HTRF detection method (cisbio).8) The obtained results were converted to activity relative to an uninhibited control (100%) and IC50 values were calculated using Prism software (GraphPad Software, Inc., U.S.A.).
Crystallization and Structural AnalysisPDE10A crystals were obtained as previously reported.30) Briefly, crystals were precipitated by sitting drop vapor diffusion methods. The reservoir solution contains 0.05 M bis-tris propane (pH 6.0), 0.1 M magnesium sulfate, 15–18% (w/v) PEG3350. Compounds were soaked into the apo PDE10A crystal. An apo crystal was transferred to a mother liquor containing compound (5 mM final concentration) and incubated at 4°C for three days. X-ray diffraction data were collected at AR-NE3A beamline31) at the Photon Factory in the National Laboratory for High Energy Physics (KEK), Tsukuba, Japan. Diffraction data were indexed, integrated, and scaled using HKL2000.32) Crystal structures of PDE10A were determined by the molecular replacement method using AMoRe33) with 2OUN30) as a search model. An initial refinement was performed using REFMAC.34) Compounds were fitted into electron densities observed in initial Fo-Fc maps using AFITT (OpenEye Scientific Software, NM, U.S.A.). Water placements and further refinements were performed using Coot35) and REFMAC, and the final models were determined. Data collection and refinement statistics are summarized in Table 4.
Table 4. Data Processing and Refinement Statistics
| Compound | 2 | 7 |
|---|
| PDB ID | 5XUJ | 5XUI |
| Space group | P212121 | P212121 |
| a (Å) | 50.3 | 49.3 |
| b (Å) | 81.3 | 81.4 |
| c (Å) | 157.6 | 158.7 |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 90 | 90 |
| Resolution (Å) | 50.0–2.44 (2.48–2.44) | 50.0–2.77 (2.82–2.77) |
| Multiplicity | 4.8 (5.0) | 5.5 (5.1) |
| Average I/σ(I) | 31.7 (5.8) | 25.2 (4.1) |
| Rmergea) (%) | 7.3 (35.6) | 8.7 (41.9) |
| No. of reflections | 21730 (1567) | 15497 (1103) |
| Completeness (%) | 92.0 (91.7) | 96.6 (98.2) |
| Rworkb) (%) | 19.8 | 18.5 |
| Rfreec) (%) | 28.0 | 27.2 |
| Average B factor (Å2) | 52.4 | 57.6 |
| RMSD bond length (Å) | 0.011 | 0.010 |
| RMSD bond angle (°) | 1.569 | 1.461 |
Values for the outer shell are given in parentheses. a) Rmerge=∑hkl ∑i|Ii−I|/∑hkl ∑iIi, where Ii is the intensity of an individual reflection and I is the mean intensity obtained from multiple observations of symmetry related reflections. b) Rwork=∑hkl||Fobs|−|Fcalc||/∑hkl|Fobs|. c) Five percent randomly omitted reflections were used for Rfree.
Pooled mouse or human liver microsomes (Xenotech LLC., U.S.A.) were diluted in 100 mM KH2PO4/K2HPO4 buffer (pH 7.4) containing 0.1 mM ethylenediaminetetraacetic acid (EDTA). The incubation mixtures (270 µL total volume), which contained 0.2 mg/mL of microsomal proteins, and 0.2 µM of substrates were pre-incubated for approximately 15 min at 37°C. Reactions were initiated by the addition of 1 mM reduced nicotinamide adenine dinucleotide phosphate (NADPH) (30 µL). After the appropriate incubation time (0, 15, 30, 45 min), 40 µL of incubation mixture was transferred into 80% acetonitrile containing internal standard (200 nM methyltestosterone, 250 µL), stood at 4°C for 20 min, and centrifuged for 20 min at 2800 rpm. The supernatant (200 µL) was prepared and analyzed via LC-MS/MS with UPLC system (Waters) and Xevo TQ (Waters). The in vitro intrinsic clearance (CLint, vitro) was calculated using Eq. 1, which is based on the time course of the residual ratio of the compounds.36)
 | (1) |
where the
Ke is the disappearance rate constant.
CYPs Inhibitory AssaysTime-dependent inhibition assay for CYP activity was performed in two steps, a pre-incubation step where the test compound was incubated with human liver microsomes and the secondary incubation period where specific substrates were added to the preincubate to measure residual CYP activity. Specific metabolites were used to monitor the CYP activities.
Each test compound (5 µM) was pre-incubated with human liver microsomes (0.1 mg/mL) and NADPH (1.0 mM) at 37°C. The pre-incubation times used were 0 and 30 min. Following the pre-incubation step, each compound was co-incubated with substrates at 37°C for 20 min. At the end of the incubation, the reaction was terminated by the addition of aqueous solution containing 80% acetonitrile. The concentration of metabolites was determined by LC-MS analysis. The inhibition of CYP activities was assessed by comparing the amount of metabolites formed in the presence of single concentration of inhibitor to the amount of metabolites formed in the solvent control. In each study, a CYP potent and specific inhibitors were used as positive control.
 | (2) |
where Activity new molecular entity (NME), 30 min is the activity in the presence of test compound and with pre-incubation, and Activity vehicle, 30 min is the activity in the absence of test compound and with pre-incubation.
The substrates of CYP1A2, 2C8, 2C9, 2C19, 2D6 and 3A4 were phenacetin (20 µM), amodiaquine (0.1 µM), diclofenac (10 µM), S-mephenytoin (30 µM), dextromethorphan (7.0 µM) and midazolam (1.5 µM), respectively.
Transcellular Transport Study in LLC-PK1-Multiple Drug Resistance 1 (MDR1) CellsWild type or MDR1-expressing LLC-PK1 cells (LLC-PK1-WT or LLC-PK1-MDR1, respectively) cultured for 5 d on a Millicell-96 Cell Culture Insert Plate (Millipore) were pre-incubated with transport buffer (HBSS, pH 7.4, for the apical and basolateral sides) for 1 h. After aspiration of the transport buffer, the donor solution (transport buffer (0.5% dimethyl sulfoxide (DMSO)) containing the test compound (1 µM) and Texas Red (1 µM)) was added to the apical or basolateral side for the influx or efflux transport study, respectively, and the receiver solution (transport buffer (0.5% DMSO)) was added to the opposite side. After incubation for 3 h, the test compound in both sides was analyzed by LC/MS/MS and the apparent permeability was determined. Efflux ratio (ER) was calculated by dividing the apparent permeability in the direction from the basolateral to the apical side by that in the opposite direction. Net efflux ratio (NER) was the ratio of ER of LLC-PK1-MDR1 to LLC-PK1-WT. Texas Red was used for the estimation of the apparent permeability via para cellular transport.