
- |<
- <
- 1
- >
- >|
-
 Akira Otaka2018Volume 66Issue 3 Pages 189-190
Akira Otaka2018Volume 66Issue 3 Pages 189-190
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
JOURNAL FREE ACCESS FULL-TEXT HTMLEditor's pickCutting-edge contributions from invited poster presenters providing significant research works in the third International Symposium for Medicinal Sciences (ISMS) in the 137th Sendai annual meeting in 2017 are assembled for the Current Topics section in this issue of the Chemical and Pharmaceutical Bulletin.
Download PDF (182K) Full view HTML
-
Yu-Shi Tian, Yi Zhou, Tatsuya Takagi, Masanori Kameoka, Norihito Kawas ...2018Volume 66Issue 3 Pages 191-206
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLThe global occurrence of viral infectious diseases poses a significant threat to human health. Dengue virus (DENV) infection is one of the most noteworthy of these infections. According to a WHO survey, approximately 400 million people are infected annually; symptoms deteriorate in approximately one percent of cases. Numerous foundational and clinical investigations on viral epidemiology, structure and function analysis, infection source and route, therapeutic targets, vaccines, and therapeutic drugs have been conducted by both academic and industrial researchers. At present, CYD-TDV or Dengvaxia® is the only approved vaccine, but potent inhibitors are currently under development. In this review, an overview of the viral life circle and the history of DENVs is presented, and the most recently reported antiviral candidates and newly discovered promising targets are focused and summarized. We believe that these successes and failures have enabled progress in anti-DENV drug discovery and hope that our review will stimulate further innovation in this area.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (4293K) Full view HTML -
Taishi Higashi, Daisuke Iohara, Keiichi Motoyama, Hidetoshi Arima2018Volume 66Issue 3 Pages 207-216
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLSupramolecular chemistry is an extremely useful and important domain for understanding pharmaceutical sciences because various physiological reactions and drug activities are based on supramolecular chemistry. However, it is not a major domain in the pharmaceutical field. In this review, we propose a new concept in pharmaceutical sciences termed “supramolecular pharmaceutical sciences,” which combines pharmaceutical sciences and supramolecular chemistry. This concept could be useful for developing new ideas, methods, hypotheses, strategies, materials, and mechanisms in pharmaceutical sciences. Herein, we focus on cyclodextrin (CyD)-based supermolecules, because CyDs have been used not only as pharmaceutical excipients or active pharmaceutical ingredients but also as components of supermolecules.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (5754K) Full view HTML -
Masayuki Kuroda, Yasushi Saito, Masayuki Aso, Koutaro Yokote2018Volume 66Issue 3 Pages 217-224
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
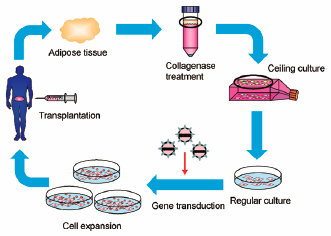
JOURNAL FREE ACCESS FULL-TEXT HTMLDespite the critical need for lifelong treatment of inherited and genetic diseases, there are no developmental efforts for most such diseases due to their rarity. Recent progress in gene therapy, including the approvals of two products (Glybera and Strimvelis) that may provide patients with sustained effects, has shed light on the development of gene therapy products. Most gene therapy products are based on either adeno-associated virus-mediated in vivo gene transfer to target tissues or administration of ex vivo gene-transduced hematopoietic cells. In such circumstances, there is room for different approaches to provide clinicians with other therapeutic options through a variety of principles based on studies not only to gain an understanding of the pathological mechanisms of diseases, but also to understand the physiological functions of target tissues and cells. In this review, we summarize recent progress in gene therapy-mediated enzyme replacement and introduce a different approach using adipocytes to enable lifelong treatment for intractable plasma protein deficiencies.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (1194K) Full view HTML -
Seiji Okada, Kulthida Vaeteewoottacharn, Ryusho Kariya2018Volume 66Issue 3 Pages 225-230
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
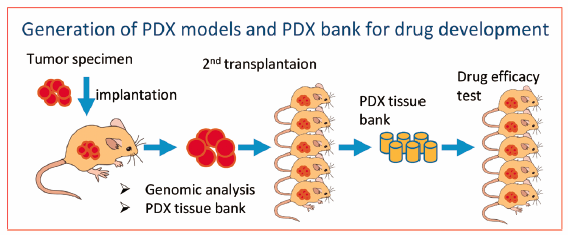
JOURNAL FREE ACCESS FULL-TEXT HTMLPatient-derived xenograft (PDX) models can be created with the transplantation of cancerous cells or tissues from patients’ primary tumors into immunodeficient mice. PDXs are now in the spotlight as more accurate human cancer models compared with mouse tumor and human cancer cell lines transplanted into mice. PDX technology leads to breakthroughs with the introduction of novel, highly immunodeficient mice such as NOG (NOD/Scid/IL2Rγnull), NSG (NOD/Scid/IL2Rγnull), and NOJ (NOD/Scid/Jak3null) mice. Xenograft efficiency differs by type of tumor, site of implantation, and tumor aggressiveness. Subcutaneous implantation is a standard method for PDX, and renal capsule or orthotropic implantation improves the efficiency. Despite positive test results in animal cancer models, significant numbers of novel drug candidates fail in clinical trials because conventional animal models such as murine tumor and human cancer cell line transplantation models do not always reflect the nature of human cancers. Since PDXs conserve the original tumor characteristics such as heterogeneous histology, clinical biomolecular signatures, malignant phenotypes and genotypes, tumor architecture, and tumor vasculature, they are currently believed to offer relevant predictive insights into clinical outcomes when evaluating the efficacy of novel cancer therapies. PDX banks with integrated genomic signatures are now established in many organizations including pharmaceutical companies. These PDX databases are becoming powerful tools for advancing precision cancer medicine.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (2559K) Full view HTML
-
Kouhei Ishizawa, Hideoki Nagai, Yohei Shimizu, Motomu Kanai2018Volume 66Issue 3 Pages 231-234
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
Advance online publication: August 19, 2017 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialA catalytic carboxylic acid-selective aldol reaction with trifluoromethyl ketones was developed. Reversible and selective covalent bond formation between a boron catalyst and a carboxylic acid is key to realizing the unprecedented catalytic aldol reaction of simple carboxylic acids. The reaction proceeded chemoselectively at the α-position of carboxylic acid even in the presence of ketone, ester, or amide functional groups in the donor substrates. The chemoselectivity is beneficial for late-stage derivatizations of biologically relevant compounds, as demonstrated by the conversion of indomethacin and triacetylcholic acid.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (506K) Full view HTML -
Tatsuya Hirano, Tomohiko Kinoshita, Daichi Kazamori, Satoshi Inoue, Ko ...2018Volume 66Issue 3 Pages 235-238
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
Advance online publication: January 11, 2018 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialWFQ-101 with a unique N-1 substituent, 5-amino-4-fluoro-2-(hydroxymethyl)phenyl group, was selected as a lead compound through combination screening based on antimicrobial activity and the efflux index against quinolone-resistant (QR) Pseudomonas aeruginosa (P. aeruginosa). Through structural optimization, we identified WFQ-228 as a novel fluoroquinolone antibiotic candidate. WFQ-228 had potent and superior activity in comparison to levofloxacin (LVX) and ciprofloxacin (CIP) against clinical isolates of P. aeruginosa, Escherichia coli and Acinetobacter baumannii, including QR strains. Furthermore, WFQ-228 demonstrated the potential to overcome major mechanisms of drug resistance; its antimicrobial activity was less affected by both pump-mediated efflux and mutations of the quinolone resistance-determining region in P. aeruginosa compared with LVX and CIP. These results suggest that WFQ-228 is a promising candidate for further evaluation in the treatment of infections caused by QR Gram-negative pathogens.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (399K) Full view HTML -
Tomokazu Ohishi, Daniel Ken Inaoka, Kiyoshi Kita, Manabu Kawada2018Volume 66Issue 3 Pages 239-242
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
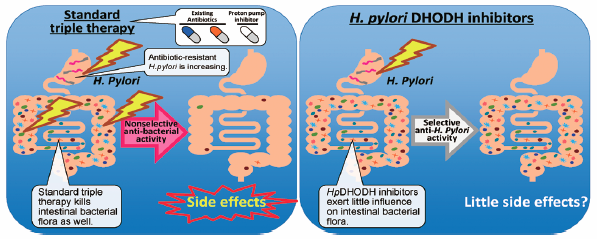
JOURNAL FREE ACCESS FULL-TEXT HTMLHelicobacter pylori (H. pylori) infection is the world’s most common bacterial infection, affecting approximately 50% of the global population. H. pylori is the strongest known risk factor for stomach diseases, including cancer. Hence, treatment for H. pylori infection can help reduce the risk of these diseases. However, the emergence of drug-resistant strains of H. pylori and the occurrence of adverse effects resulting from current therapies have complicated the successful eradication of H. pylori infection. Although various antibiotics that target several bacterial enzymes have been discovered, dihydroorotate dehydrogenase (DHODH) may hold potential for the development of novel anti-H. pylori agents with reduced toxicity and side effects. Here we review the existing literature that has focused on strategies for developing novel therapeutic agents that target the DHODH of H. pylori.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (570K) Full view HTML
-
Yoichi Kadoh, Haruko Miyoshi, Takehiko Matsumura, Yoshihito Tanaka, Mi ...2018Volume 66Issue 3 Pages 243-250
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLPhosphodiesterase (PDE) 10A is a dual hydrolase of cAMP and cGMP and highly expressed in striatal medium spiny neurons. Inhibition of PDE10A modulates the activity of medium spiny neurons (MSN) via the regulation of cAMP and cGMP. Signal control of MSN is considered associated with psychotic symptoms. Therefore PDE10A inhibitor is expected as a therapeutic method for psychosis disease such as schizophrenia. Avanafil (1) is a PDE5 inhibitor (treatment for erectile dysfunction) discovered by our company. We paid attention to the homology of PDE10A and PDE5 and took advantage of PDE5 inhibitor library to discover PDE10A inhibitors, and found a series of compounds that exhibit higher potency for PDE10A than PDE5. We transformed the afforded derivatives, which had weak inhibitory activity against PDE10A, and discovered stilbene as a PDE10A inhibitor. Brain penetration of this compound was improved by further conversion of N-containing heterocycles and their substituents. The afforded dimethylaminopyrimidine was effective for rat conditioned avoidance response (CAR) test; however, it did not exhibit good brain penetration. We performed in-depth optimization focusing on substituents of the quinoxaline ring, and produced 3-methyl-7-fluoro quinoxaline. This compound was the most effective in rat CAR test due to its strong PDE10A inhibitory activity and good pharmacokinetics.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (1066K) Full view HTML -
Kazuhiko Iikubo, Yutaka Kondoh, Itsuro Shimada, Takahiro Matsuya, Keni ...2018Volume 66Issue 3 Pages 251-262
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLAnaplastic lymphoma kinase (ALK) is a validated therapeutic target for treating echinoderm microtubule-associated protein-like 4 (EML4)-ALK positive non-small cell lung cancer (NSCLC). We synthesized a series of 1,3,5-triazine derivatives and identified ASP3026 (14a) as a potent and selective ALK inhibitor. In mice xenografted with NCI-H2228 cells expressing EML4-ALK, once-daily oral administration of 14a demonstrated dose-dependent antitumor activity. Here, syntheses and structure–activity relationship (SAR) studies of 1,3,5-triazine derivatives are described.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (3177K) Full view HTML
-
Taichiro Togo, Toshiya Taniguchi, Yoshitaka Nakata2018Volume 66Issue 3 Pages 263-269
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLAripiprazole (APZ) is used to treat schizophrenia and is administered as a tablet containing the anhydrous form of APZ. In this study, the effect of compaction force on the crystal form transition was investigated. The crystalline state was observed by X-ray diffraction (XRD). APZ Anhydrous Form II was compacted into tablets. The XRD intensity of anhydrous APZ became lower with higher compressive force. The degree of crystallinity decreased with the compaction force. The powder and the compacted tablets of anhydrous APZ were stored for one week under 60°C and 75% relative humidity. The powder showed no crystal form transition after storage. For the tablets, however, XRD peaks of APZ hydrate were observed after storage. The tablets compacted with higher force showed the higher XRD diffraction intensity of hydrate form. We concluded that the crystallinity reduction of APZ Anhydrous Form II by compaction caused and accelerated the transition to hydrate under high temperature and humidity conditions. In order to manufacture crystallographically stable tablets containing anhydrous APZ, it is important to prevent this crystallinity reduction during compaction.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (1093K) Full view HTML -
Yo Muraki, Midori Yamasaki, Hirohisa Takeuchi, Kimio Tohyama, Noriyasu ...2018Volume 66Issue 3 Pages 270-276
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
Advance online publication: January 06, 2018 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLPulmonary hypertension (PH) is a life-threatening lung disease. Despite the availability of several approved drugs, the development of a new treatment method is needed because of poor prognosis. Tissue selective drug delivery systems can avoid the adverse effects of current therapy and enhance efficacy. We evaluated the possibility of delivering drugs to the lungs of a PH rat model using fluorescence dye-labeled nanosized liposomes. To evaluate the tissue distribution following systemic exposure, fluorescent dye-labeled, 40–180 nm liposomes with and without polyethylene glycol (PEG) were intravenously administered to a monocrotaline-induced PH (MCT) rat model and tissue fluorescence was measured. Fluorescent dye-containing liposomes were intratracheally administered to the MCT model to evaluate the distribution of the liposome-encapsulated compound following local administration to reduce systemic exposure. The lung vascular permeability, plasma concentration of surfactant protein (SP)-D, lung reactive oxygen species (ROS) production, and macrophage marker gene cluster of differentiation (CD68) expression were measured. PEG and 80-nm liposome accumulation in the lung was elevated in the MCT model compared to that in normal rats. The intratracheally administered liposomes were delivered selectively to the lungs of the MCT model. The lung vascular permeability, plasma SP-D concentration, and CD68 expression were significantly elevated in the lungs of the MCT model, and were all significantly and positively correlated to liposome lung accumulation. Liposomes can accumulate in the lungs of an MCT model by enhancing vascular permeability by the inflammatory response. Therefore, drug encapsulation in liposomes could be an effective method of drug delivery in patients with PH.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (2456K) Full view HTML -
Khaled Mohamed Elamin, Yuki Yamashita, Taishi Higashi, Keiichi Motoyam ...2018Volume 66Issue 3 Pages 277-285
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
Advance online publication: December 22, 2017 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLMethyl-β-cyclodextrin (M-β-CyD) exhibits cytotoxic activity, and has the potentials as an antitumor agent. However, a tumor-selectivity of M-β-CyD is low, leading to low antitumor activity and the adverse effects. Meanwhile, hyaluronic acid (HA) is known as a promising tumor targeting ligand, because various cancer cells overexpress CD44, a HA-binding glycoprotein. In the present study, to develop a tumor-selective delivery system for M-β-CyD, we designed a supramolecular complex of M-β-CyD with adamantane-grafted HA (Ad-HA/M-β-CyD) and evaluated it as a tumor-selective antitumor agent. M-β-CyD formed a stable complex with Ad-HA (Kc>104 M−1). In addition, Ad-HA/M-β-CyD formed slightly a negative-charged nanoparticle with ca. 140 nm of a particle size, indicating the favorable physicochemical properties for antitumor agents. Ad-HA/M-β-CyD showed the superior cytotoxic activity via CD44-mediated endosomal pathways in HCT116 cells (CD44(+)), a human colon cancer cell line. In addition, cytotoxic activity of Ad-HA/M-β-CyD was induced by apoptosis. These results suggest that Ad-HA/M-β-CyD has the potentials as a tumor-selective supramolecular antitumor agent.
Graphical Abstract Fullsize ImageView full abstractEditor's pickMethyl-b-cyclodextrin (M-b-CyD) exhibits an antitumor activity, although its tumor-selectivity is low. Meanwhile, hyaluronic acid (HA) is known as a promising tumor targeting ligand, because various cancer cells overexpress CD44, a HA-binding glycoprotein. In this study, to develop a tumor-selective delivery system for M-b-CyD, the authors designed a supramolecular complex of M-b-CyD with adamantane-grafted HA (Ad-HA/M-b-CyD) and evaluated it as a tumor-selective antitumor agent. Eventually, Ad-HA/M-b-CyD showed the superior cytotoxic activity via CD44-mediated endosomal pathways, suggesting that Ad-HA/M-b-CyD has the potentials as a tumor-selective antitumor agent.
Download PDF (1840K) Full view HTML -
Ayaka Chino, Ryushi Seo, Yasushi Amano, Ichiji Namatame, Wataru Hamagu ...2018Volume 66Issue 3 Pages 286-294
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLIn this study, we report the identification of potent pyrimidoindazoles as phosphodiesterase10A (PDE10A) inhibitors by using the method of fragment-based drug discovery (FBDD). The pyrazolopyridine derivative 2 was found to be a fragment hit compound which could occupy a part of the binding site of PDE10A enzyme by using the method of the X-ray co-crystal structure analysis. On the basis of the crystal structure of compound 2 and PDE10A protein, a number of compounds were synthesized and evaluated, by means of structure–activity relationship (SAR) studies, which culminated in the discovery of a novel pyrimidoindazole derivative 13 having good physicochemical properties.
Graphical Abstract Fullsize ImageView full abstractEditor's pickThis article describes the discovery of potent pyrimidoindazoles as PDE10A inhibitors by using the method of fragment-based drug discovery (FBDD). The authors made use of X-ray analysis well to obtain PDE10A inhibitors. In addition, they designed well-balanced compounds in light of physicochemical property and inhibitory activity. This study showed that FBDD approach is effective for the discovery of PDE10A inhibitors.
Download PDF (2304K) Full view HTML -
Fumihiko Ogata, Noriaki Nagai, Erimi Ueta, Takehiro Nakamura, Naohito ...2018Volume 66Issue 3 Pages 295-302
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLIn this study, we prepared novel adsorbents containing virgin and calcined tapioca products for removing strontium (Sr(II)) and cesium (Cs(I)) from aqueous solutions. The characteristics of tapioca, along with its capacity to adsorb Sr(II) and Cs(I), were evaluated. Multiple tapioca products were prepared and tested. The adsorbent prepared by boiling the tapioca followed by calcination at 300°C (BTP300) was the most effective. In addition, adsorption was affected by the adsorbent’s surface properties. The Sr(II) and Cs(I) adsorbed onto BTP300 could be recovered through desorption by hydrochloric acid at different concentrations, which indicates that BTP300 can be used several times for adsorption/desorption. The results of this study suggest that BTP300, which was produced from tapioca biomass, can remove Sr(II) and Cs(I) from aqueous solutions.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (2943K) Full view HTML -
Yukiko Karuo, Kohei Yamada, Munetaka Kunishima2018Volume 66Issue 3 Pages 303-308
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
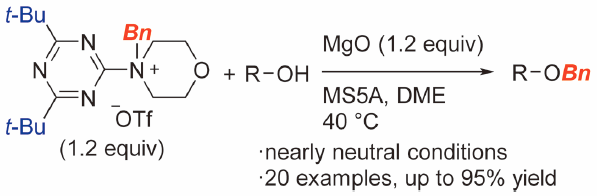
Supplementary materialBenzylating reagents, 4-(4,6-di-t-butyl-1,3,5-triazin-2-yl)-4-benzylmorpholinium triflate, and related derivatives have been developed. The reagents release benzyl triflate as a benzyl cation equivalent upon heating the solution to 40°C under neutral conditions. The O-benzylation of alcohols using a stoichiometric amount of these reagents afforded corresponding benzyl ethers in good to high yields. This was due to the presence of a bulky t-butyl group on the triazine ring of these reagents that prevents the consumption of benzyl triflate via a side reaction with a morpholinotriazine derivative.
Graphical Abstract Fullsize ImageView full abstractEditor's pickThis paper describes the development of triazine-based benzylating reagents, 4-(4,6-di-t-butyl-1,3,5-triazin-2-yl)-4-benzylmorpholinium triflate, and related derivatives. These reagents generate benzyl triflate (BnOTf) as a benzyl cation equivalent upon heating the solution to 40 °C under neutral conditions. The incorporation of a bulky t-butyl group into the triazine ring of the reagents prevents the formation of N-benzyltriaziniums, which are byproducts from the reaction of BnOTf and co-products, morpholinotriazine derivatives. O-Benzylation of various alcohols with a stoichiometric amount of these reagents proceeded in good yields.
Download PDF (856K) Full view HTML -
Rafat Milad Mohareb, Amira Elsayed Mahmoud Abdallah, Abeer Abdelazeem ...2018Volume 66Issue 3 Pages 309-318
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
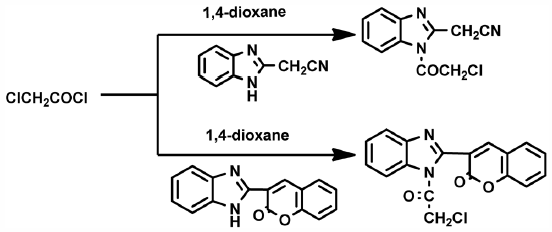 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLThe reactivity of compounds 2-(1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)acetonitrile 2 and 3-(1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)-2H-chromen-2-one 8 towards different chemical reagents were studied and a series of novel benzimidazole derivatives were obtained (2–6a–d and 8–12a–d). Moreover, in vitro growth inhibitory effect of the newly synthesized compounds were evaluated in term of [IC50 µM] against the six cancer cell lines, human lung carcinoma (A549), lung cancer (H460), human colorectal (HT29), gasteric cancer cell (MKN-45), glioma cell line (U87MG) and cellosaurus cell line (SMMC-7721) where foretinib was used as standard reference. The results showed that compounds 2 (only for A549 cell line), 3a, 4, 6c, 6d, 8, 9a, 9e and 9f were the most active compounds towards the six cancer cell lines. On the other hand, the toxicity of these most potent compounds against shrimp larvae indicated that compounds 3a, 4, 6d, 9e and 9f were non toxic while compounds 6c and 8 were very toxic and compounds 2 and 9a were harmful against the tested organisms.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (782K) Full view HTML -
Shuoqian Liu, Jorge Freire da Silva Ferreira, Dongming Tian, Yuwei Tan ...2018Volume 66Issue 3 Pages 319-326
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
Advance online publication: December 28, 2017 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLIn order to make full use of artemisinin production waste and thus to reduce the production cost of artemisinin, we developed an efficient and scalable method to isolate high-purity dihydroartemisinic acid from artemisinin production waste by combining anion-exchange resin with silica-gel column chromatography. The adsorption and desorption characteristics of dihydroartemisinic acid on 10 types of anion-exchange resin were investigated, and the results showed that the 717 anion-exchange resin exhibited the highest capacity of adsorption and desorption to dihydroartemisinic acid. Adsorption isotherms were established for the 717 anion-exchange resin and they fitted well with both Langmuir and Freundlich model. Dynamic adsorption and desorption properties of 717 anion-exchange resin were characterized to optimize the chromatographic conditions. Subsequently, the silica-gel column chromatography was performed and dihydroartemisinic acid with a purity of up to 98% (w/w) was obtained. Finally, the scale-up experiments validated the preparative separation of high-purity dihydroartemisinic acid from industrial waste developed in the present work. This work presented for the first time an isolation of dihydroartemisinic acid with a purity of 98% from Artemisia annua (A. annua) by-product, which adds more value to this crop and has the potential to lower the prices of anti-malarial drugs.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (865K) Full view HTML -
Sabrina Dahlizar, Mika Futaki, Akie Okada, Wesam Radhi Kadhum, Hiroaki ...2018Volume 66Issue 3 Pages 327-333
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
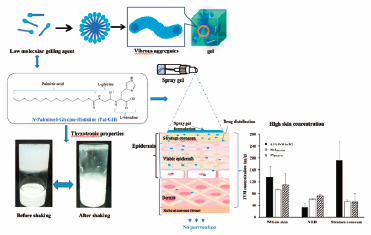 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLPalmitoyl-glycine-histidine (Pal-GH) is a new low molecular weight gelling agent. It exhibits thixotropic behavior, low viscosity, and high dissolving properties for a wide range of hydrophilic to lipophilic drugs. Orally administered ivermectin (IVM) is used to treat scabies. However, this treatment is associated with well-known side effects, thus a study is awaited to search for alternative routes of administration. Although a topical formulation of IVM could be a candidate, it requires whole body application except the head and face for several hours on a daily basis. Therefore, in this study, we prepared a gel spray formulation containing IVM as an approach for application to large skin areas with a single spray application without further contact with the applied formulation. Pal-GH gel spray formulations were prepared from its aqueous solution by a heating and cooling method. Rheological behavior and physical appearance (spraying, spreading ability, volume of spraying, and homogeneity) of the prepared formulations were evaluated. Pal-GH gel with propylene glycol demonstrated impressive rheological properties (typical thixotropic behavior) with high hysteresis area among all the tested Pal-GH gels and spreading ability. The obtained IVM concentration in the skin after topical application of 0.1% IVM-containing Pal-GH formulation onto hairless rats was much higher than the reported therapeutic concentration obtained from oral administration in humans. These results suggested that topical application of IVM using a Pal-GH gel spray formulation could be an alternative to the conventional oral forms for the scabies treatment.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (803K) Full view HTML
-
Toshiya Nagai, Kengo Hanaya, Shuhei Higashibayashi, Mitsuru Shoji, Tak ...2018Volume 66Issue 3 Pages 334-338
Published: March 01, 2018
Released on J-STAGE: March 01, 2018
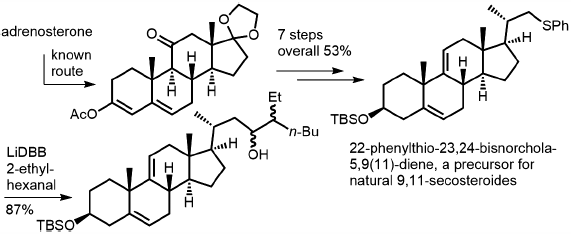 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML3β-tert-Butyldimethylsiloxy-22-phenylthio-23,24-bisnorchola-5,9(11)-diene, which has a double bond between C-9 and C-11 and a phenylsulfenyl group on the terminus of the side chain, is a potential synthetic intermediate for steroids with 9,11-unsaturation or 9,11-seco skeletons. We describe here the synthesis of the title compound from 17-ethylenedioxy-3-acetoxyandrosta-3,5-dien-11-one. The introduction of an ethylene unit to 3β-tert-butyldimethylsiloxyandrosta-5,9(11)-dien-17-one by the action of ethyltriphenylphosphonium bromide under basic conditions resulted in an inseparable mixture of two stereoisomeric products (5 : 1). However, in the subsequent step, only the (Z)-isomer was susceptible to the Lewis acid-catalyzed ene reaction with formaldehyde, giving a stereochemically pure product with the desired configuration. Within three steps, the ene-product was derivatized to the title compound, with a total yield of 53% over seven steps. Reductive terminal anion formation by treatment with lithium di-tert-butylbiphenyl (LiDBB) and subsequent nucleophilic attack on a branched aliphatic aldehyde was demonstrated, with an eye toward the introduction of side chains, especially for steroids with oxygen functionality at C-23.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (548K) Full view HTML
- |<
- <
- 1
- >
- >|