Regular Articles
Synthesis of Novel Thiophene, Thiazole and Coumarin Derivatives Based on Benzimidazole Nucleus and Their Cytotoxicity and Toxicity Evaluations
2018 Volume 66 Issue 3 Pages 309-318
Details
2018 Volume 66 Issue 3 Pages 309-318
The reactivity of compounds 2-(1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)acetonitrile 2 and 3-(1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)-2H-chromen-2-one 8 towards different chemical reagents were studied and a series of novel benzimidazole derivatives were obtained (2–6a–d and 8–12a–d). Moreover, in vitro growth inhibitory effect of the newly synthesized compounds were evaluated in term of [IC50 µM] against the six cancer cell lines, human lung carcinoma (A549), lung cancer (H460), human colorectal (HT29), gasteric cancer cell (MKN-45), glioma cell line (U87MG) and cellosaurus cell line (SMMC-7721) where foretinib was used as standard reference. The results showed that compounds 2 (only for A549 cell line), 3a, 4, 6c, 6d, 8, 9a, 9e and 9f were the most active compounds towards the six cancer cell lines. On the other hand, the toxicity of these most potent compounds against shrimp larvae indicated that compounds 3a, 4, 6d, 9e and 9f were non toxic while compounds 6c and 8 were very toxic and compounds 2 and 9a were harmful against the tested organisms.
Benzimidazole derivatives occupy an important position in medicinal chemistry and are of particular interest in the search for new bioactive compounds in the pharmaceutical industry. Thus, imidazole and benzimidazole scaffolds are extremely versatile and have featured a number of clinically used drugs such as antihistaminic,1,2) antiulcer,3) antihypertensive,4) antibacterial,5) antifungal,6,7) anti-parasitic,8) anti-emetic,9) anticancer,10) antiviral11) and other therapeutic agents with high therapeutic potency and market value.12) In the present work, a number of benzimidazole derivatives were synthesized and evaluated their biological activity as anticancer agents. The chemical structures of some of the benzimidazole based anti-cancer drugs which are candidates in different stages of clinical trials by various pharmaceutical companies are illustrated below (Fig. 1) such as AT9283,13) Galeterone14) and Veliparib.15) On the other hand, there are various methods lead to prepare benzimidazole derivatives such as a one-pot, multicomponent reaction,16) one-pot condensation reaction,17) addition reactions,18) intramolecular N-arylations reaction,19) a copper-catalyzed, one-pot and three-component reaction.20) In the present work we used the 2-(1H-benzo[d]imidazol-2-yl)acetonitrile 1 as the key starting compound for the synthesis of 1-α-chloroacetyl derivative followed by several heterocyclization to afford potentially cytotoxic derivatives.

Benzimidazole structural motifs have attracted much interest in diverse areas of medicinal chemistry. These heterocycles have shown various pharmacological activities such as anti-human immunodeficiency virus (HIV),21) poli(ADP-ribose) phosphorylase inhibitors,22) Histamine H4 receptor binders23) and cardiovascular.24) In view of the tremendous biological activities of benzimidazoles, their preparation has gained considerable attention in recent years. In view of the above-mentioned facts and in continuation of our interest in the synthesis of heterocycles containing benzimidazole moiety,25) to identify new candidates that may be value in designing new, potent, selective and less toxic anti-tumor agents, we report herein the synthesis and anti-tumor evaluation of some novel structure heterocycles incorporating benzo[d]imidazole moiety. For these reasons, we focused our effort in this work through the uses of the 2-(1H-benzo[d]imidazol-2-yl)acetonitrile 1 as the key starting compound. The synthetic strategies adopted for the synthesis of the intermediates and target compounds are depicted in Charts 1–4. Compound 1 was obtained through the reaction of o-phenylenediamine with ethyl cyanoacetate at 120°C. It was reacted with chloroacetylchloride in 1,4-dioxane to give the 2-(1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)acetonitrile 2. Structure of compound 2 was established on the basis of the obtained analytical and spectral data. Thus, the MS showed molecular ion peak [M+] (m/z 233) corresponding to molecular formula C11H8N3OCl and 1H-NMR spectrum showed two singlets at δ 3.55 and 4.38 ppm for the two CH2 groups and a multiplet at δ 7.25–7.54 ppm corresponding to the C6H4 group. In addition 13C-NMR spectrum exhibited signals at δ 14.5, 46.1 indicating the two CH2 groups, a signal at δ 119.5 eq to the CN group, signals at δ 112.1, 113.0, 123.5, 123.7, 130.4, 138.0 eq to the benzene ring, a signal at δ 164.1 eq to the C=O group and a signal at δ 174.0 corresponding to the C=N imidazole.
The reaction of compound 2 with either of hydrazine hydrate or phenylhydrazine gave the hydrazine derivatives 3a and 3b, respectively. On the other hand, treatment of compound 2 with potassium cyanide at 60°C afforded the 3-(2-(cyanomethyl)-1H-benzo[d]imidazol-1-yl)-3-oxopropanenitrile (4). The structure of the latter compound was elucidated on the basis of their spectral data (IR, MS and 1H-NMR). In addition, the reaction of compound 2 with the 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile gave the 2-((2-oxoacetyl)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene derivative 5 (Chart 1).

Recently, our research group was directed to study the reaction of active methylene reagents with phenylisothiocyanate in basic dimethylformamide/potassium hydroxide (DMF/KOH) solution followed by hetero-cyclization of the intermediate potassium sulphide salt with α-halocarbonyl compounds.26,27) The reaction gave either thiophene or thiazole derivatives depending on the nature of the used active methylene reagent and the α-halocarbonyl compound. In continuation of this program, we studied the reactivity of compound 2 as an α-halocarbonyl derivative to form thiazole derivatives (Chart 2). Thus, any of the malononitrile, ethyl cyanoacetate, acetylacetone or ethyl acetoacetate reacted with phenyl isothiocyanate in DMF/KOH solution form the intermediate potassium sulphide salt. The reaction of the latter compound with compound 2 forms the thiazole derivatives 6a–d, respectively (Chart 2). The analytical and spectral data of compounds 6a–d were consistent with their respective structures. Thus, the 1H-NMR spectrum of compound 6a (as an example) revealed the presence of a singlet at δ 4.35 ppm for the CH2 group, a singlet at range of δ 6.40 ppm for CH thiazole and a multiplet at δ 7.15–7.63 ppm for the C6H4 and C6H5 groups.
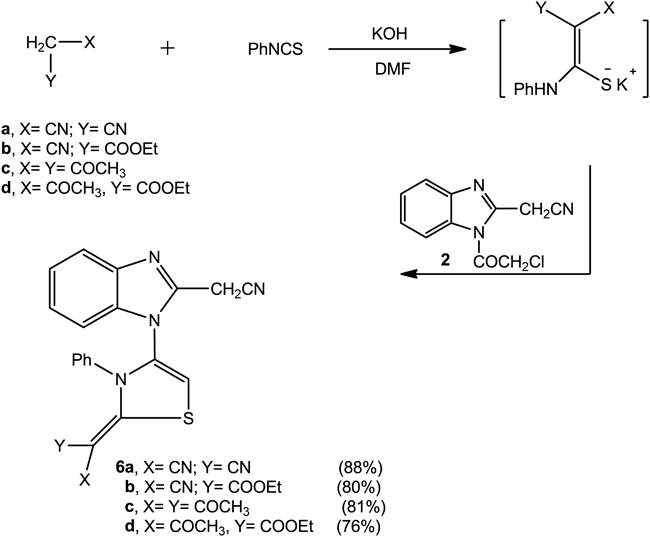
Next, the reaction of compound 1 with salicylaldehyde afforded the coumarin derivative 7. The reaction of compound 7 with chloroacetylchloride gave the N-chloroacetamido derivative 8. The latter compound showed reactivities toward some chemical reagents to form bioactive molecules. Thus, the reaction of compound 8 with any of the diazonium salts namely benzenediazonium chloride, 4-chlorobenzenediazonium chloride, 4-methoxybenzenediazonium chloride, 4-tolyldiazonium chloride, 2,4-dichlorobenzene-diazonium chloride or 4-nitrobenzenediazonium chloride afforded the hydrazonyl chloride derivatives 9a–f, respectively. On the other hand, the nucleophilic displacement of compound 8 with potassium cyanide gave the N-cyanoacetyl derivative 10 (Chart 3).
Compound 2 reacted with thioglycollic acid to give the thiazole derivative 11, its structure was confirmed on the basis of analytical and spectral data. The 1H-NMR spectrum showed three singlets at δ 3.38, 3.70 and 4.38 ppm for the three CH2 groups and a multiplet at δ 7.26–7.55 ppm for C6H4 moiety.

Multi-component reaction (MCR) is a procedure wherein three or more, commercially accessible or easily available components are gathered together through one pot method to provide an expected target; demonstrating features of starting materials, hence provide better chances for molecular diversity per step in short reaction time and attaining better yields. With a narrow set of starting precursors, a wide range of libraries can be formed in a short reaction time through MCRs.28–32) It is quite remarkable that many top-selling pharmaceuticals contain 4H-pyran derivatives33–36) encouraged us to synthesis 4H-pyran derivative through the MCRs of compound 11. Thus, the reaction of compound 11 with any of the aromatic aldehydes like benzaldehyde, 4-chlorobenzaldehyde, 4-methoxybenzaldehyde or 2-nitrobenzaldehyde gave the pyran derivatives 12a–d, respectively (Chart 4). The structures of compounds 12a–d were established on the basis of their respective analytical and spectral data (see Experimental).

The anti-proliferative activities of the newly synthesized compounds (Table 1) were evaluated against the six cancer cell lines, human lung carcinoma (A549), human colorectal (HT29), gasteric cancer cell (MKN-45), glioma cell line (U87MG), and cellosaurus cell line (SMMC-7721) and lung cancer (H460) using the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in vitro, with foretinib as the positive control.37–39) The cancer cell lines were cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS). Approximate 4×103 cells, suspended in MEM medium, were plated onto each well of a 96-well plate and incubated in 5% CO2 at 37°C for 24 h. The compounds tested at the indicated final concentrations were added to the culture medium and the cell cultures were continued for 72 h. Fresh MTT was added to each well at a terminal concentration 5 µg/mL, and incubated with cells at 37°C for 4 h. The formazan crystals were dissolved in 100 µL of dimethyl sulfoxide (DMSO) each well, and the absorbency at 492 nM (for absorbance of MTT formazan) and 630 nM (for the reference wavelength) was measured with an enzyme-linked immunosorbent assay (ELISA) reader. All of the compounds were tested three times in each cell line. The results expressed as half maximal inhibitory concentration (IC50) were the averages of three determinations and calculated by using the Bacus Laboratories Incorporated Slide Scanner (Bliss) software. The mean values of three independent experiments, expressed as IC50 values, were presented in Table 1. Most of the synthesized compounds exhibited potent anti-proliferative activity with IC50 values less than 30 µM. Generally, the variations of substituents within the thienopyridine moiety together with the heterocycle ring being attached have a notable influence on the anti-proliferative activity.
| Compound No. | IC50±S.E.M. (µM)a) | |||||
|---|---|---|---|---|---|---|
| A549 | H460 | HT29 | MKN-45 | U87MG | SMMC-7721 | |
| 2 | 0.80±0.12 | 9.59±2.83 | 8.48±3.21 | 6.49±2.49 | 8.66±2.52 | 7.27±2.97 |
| 3a | 0.38±0.09 | 0.42±0.09 | 0.29±0.07 | 0.88±0.29 | 1.83±0.29 | 0.72±0.09 |
| 3b | 6.29±2.04 | 8.80±3.05 | 6.28±2.36 | 5.77±2.18 | 8.69±2.59 | 6.32±1.52 |
| 4 | 0.83±0.26 | 0.39±0.05 | 0.61±0.27 | 0.80±0.36 | 0.36±0.46 | 1.53±0.29 |
| 5 | 8.29±2.33 | 8.42±2.29 | 9.18±1.44 | 6.62±1.93 | 8.58±2.69 | 8.51±2.64 |
| 6a | 8.36±4.69 | 2.66±1.58 | 8.73±2.66 | 6.83±2.69 | 8.40±2.59 | 4.66±1.50 |
| 6b | 8.28±3.69 | 7.29±2.64 | 8.59±4.39 | 9.38±2.72 | 9.42±2.93 | 6.36±2.82 |
| 6c | 1.55±0.49 | 2.56±0.52 | 0.49±0.16 | 0.94±0.59 | 3.28±0.63 | 0.25±0.64 |
| 6d | 0.38±0.05 | 0.53±0.04 | 0.66±0.04 | 0.23±0.06 | 0.82±0.14 | 0.49±0.09 |
| 7 | 6.63±2.69 | 8.79±2.39 | 6.70±1.41 | 2.90±0.93 | 1.69±0.72 | 2.59±0.83 |
| 8 | 0.29±0.03 | 0.59±0.28 | 0.57±0.08 | 0.80±0.39 | 0.34±0.09 | 0.69±0.40 |
| 9a | 0.48±0.23 | 0.70±0.58 | 0.69±0.42 | 0.66±0.39 | 1.27±0.74 | 0.59±0.32 |
| 9b | 8.87±2.69 | 6.21±1.42 | 8.69±1.69 | 9.69±2.57 | 9.28±2.55 | 6.59±2.38 |
| 9c | 8.66±2.29 | 9.29±1.97 | 8.73±2.79 | 6.63±2.68 | 6.28±0.39 | 8.93±2.52 |
| 9d | 8.28±3.27 | 8.60±2.80 | 1.35±0.59 | 1.43±0.61 | 10.04±3.17 | 10.79±4.51 |
| 9e | 0.25±0.19 | 0.36±0.16 | 0.89±0.27 | 0.73±0.06 | 0.29±0.08 | 0.22±0.05 |
| 9f | 0.28±0.04 | 0.61±0.49 | 0.88±0.33 | 1.38±0.59 | 3.33±0.06 | 2.52±0.16 |
| 10 | 8.31±1.88 | 9.21±2.60 | 7.42±1.09 | 9.31±2.72 | 8.59±2.97 | 8.53±2.29 |
| 11 | 2.39±0.85 | 1.08±0.69 | 2.68±0.86 | 8.69±2.03 | 8.48±2.93 | 9.59±3.49 |
| 12a | 6.23±1.25 | 6.09±1.27 | 9.58±1.59 | 8.32±2.42 | 8.22±1.49 | 7.58±2.83 |
| 12b | 1.26±0.84 | 1.75±0.32 | 8.59±2.73 | 4.68±1.48 | 9.45±2.58 | 7.29±2.19 |
| 12c | 8.35±2.69 | 8.53±3.70 | 7.84±2.69 | 6.38±1.42 | 8.68±2.80 | 8.48±1.69 |
| 12d | 9.47±0.14 | 8.30±1.69 | 9.08±1.58 | 7.34±2.48 | 9.20±1.84 | 6.22±2.80 |
| Foretinib | 0.08±0.01 | 0.18±0.03 | 0.15±0.023 | 0.03±0.0055 | 0.90±0.13 | 0.44±0.062 |
a) Data are expressed as means±S.E.M. of three independent experiments performed in duplicates.
It is clear from Table 1 that most of the synthesized compounds showed from moderate to high potency against the six cancer cell lines. Compound 2 showed high cytotoxicity only against A549 with IC50 0.80 µM. Moreover, compound 3a with (R=H) showed higher cytotoxicity than 3b (R=Ph). It is worthy to mention that the high nitrogen content in compound 3a was responsible for its high potency. Compound 4 showed high cytotoxicity against the six cancer cell lines. It is obvious that the CN group present in compound 4 was responsible for it high activity. The reaction of compound 2 with the 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene to give compound 5, surprising with moderate cytotoxicity against the six cancer cell lines. Considering the thiazole derivatives 6a–d it is obvious that compounds 6c (X=Y=COCH3) and 6d (X=COCH3, Y=COOEt) showed most activities among such series of compounds. It is very common between compounds 6c and 6d that the presence of the COCH3 was responsible for the high potency of these two compounds. Moreover, the presence of the high oxygen content COOEt moiety was responsible for the extremely high potency of compound 6d. In addition, compound 6c showed higher potency than the reference foretinib against the SMMC-7721 cell line. On the other hand, the coumarin derivative 7 showed moderate cytotoxicity. Moreover, alkylation of compound 7 to give compound 8 showed high potency against the six cancer cell lines. Such reactivity of compound 8 was attributed to the presence of the COCH2Cl moiety. Considering the hydrazidic halide derivatives 9a–f, compounds 9a (X=Y=H), 9e (X=Y=Cl) and 9f (X=NO2, Y=H) showed high cytotoxicity against the six cancer cell lines. Very common between compounds 9a–c the presence of the –NH–N=C–(CO)Cl moiety which was responsible of their high potencies. In addition the presence of the two Cl groups in 9e and the NO2 group in 9f extremely increase their potencies. It was surprisingly that compound 10, which was produced through the nucleophilic displacement of the Cl in compound 8 into CN showed moderate potencies towards the six cancer cell lines. Similarly, compounds 11 and 12a–d showed moderate potency, although compound 12b showed slight activities against A549 and H460 cell lines with IC50’s 1.26 and 1.75 µM. The cytotoxicity evaluation of the most active newly synthesized compounds toward the six cancer cell lines are illustrated through Fig. 2.

Bioactive compounds are often toxic to shrimp larvae. Thus, in order to monitor these chemicals in vivo lethality to shrimp larvae (Artemia salina), Brine-Shrimp Lethality Assay40) was used. Results were analyzed with LC50 program to determine LC50 values and 95% confidence intervals.41,42) Results are given in Table 2 for the compounds which exhibited optimal cytotoxic effect against the cancer cell lines which are the nine compounds 2, 3a, 4, 6c, 6d, 8, 9a, 9e and 9f.
The shrimp lethality assay is considered as a useful tool for preliminary assessment of toxicity, and it has been used for the detection of fungal toxins, plant extract toxicity, heavy metals, cyanobacteria toxins, pesticides, and cytotoxicity testing of dental materials.43) It has also been shown that, A. salina toxicity test results have a correlation with rodent and human acute oral toxicity data. Generally, a good correlation was obtained between A. salina toxicity test and the rodent data. Likewise, the predictive screening potential of the aquatic invertebrate tests for acute oral toxicity in man, including A. salina toxicity test, was slightly better than the rat test for test compounds.
In order to prevent the toxicity results from possible false effects originated from solubility of compounds and DMSO’s possible toxic effect, the tested compounds were prepared by dissolving in DMSO in the suggested DMSO volume ranges. It is clear from Table 2 that compounds 3a, 4, 6d, 9e and 9f showed non toxicity against the tested organisms. On the other hand, compounds 6c and 8 were very toxic; in addition, compounds 2, 9a were harmful.
| Compound No. | Cons. (µg/mL) | Mortalitya) | Toxicity | LC50 | Upper 95% lim | Lower 95% lim |
|---|---|---|---|---|---|---|
| 2 | 10 | 0 | Harmful | 119.35 | 70.38 | 26.79 |
| 100 | 5 | |||||
| 1000 | 10 | |||||
| 3a | 10 | 0 | Non toxic | 972.31 | — | — |
| 100 | 0 | |||||
| 1000 | 4 | |||||
| 4 | 10 | 0 | Non toxic | 860.20 | — | — |
| 100 | 2 | |||||
| 1000 | 4 | |||||
| 6c | 10 | 5 | Very toxic | 138.32 | — | — |
| 100 | 8 | |||||
| 1000 | 10 | |||||
| 6d | 10 | 2 | Non toxic | 890.11 | — | — |
| 100 | 6 | |||||
| 1000 | 10 | |||||
| 8 | 10 | 2 | Very toxic | 44.8 | — | — |
| 100 | 6 | |||||
| 1000 | 10 | |||||
| 9a | 10 | 0 | Harmful | 140.29 | 70.40 | 18.90 |
| 100 | 3 | |||||
| 1000 | 8 | |||||
| 9e | 10 | 0 | Non toxic | 888.28 | — | — |
| 100 | 2 | |||||
| 1000 | 4 | |||||
| 9f | 10 | 0 | Non-toxic | 856.28 | — | — |
| 100 | 0 | |||||
| 1000 | 4 |
a) Ten organisms (A. salina) tested for each concentration. 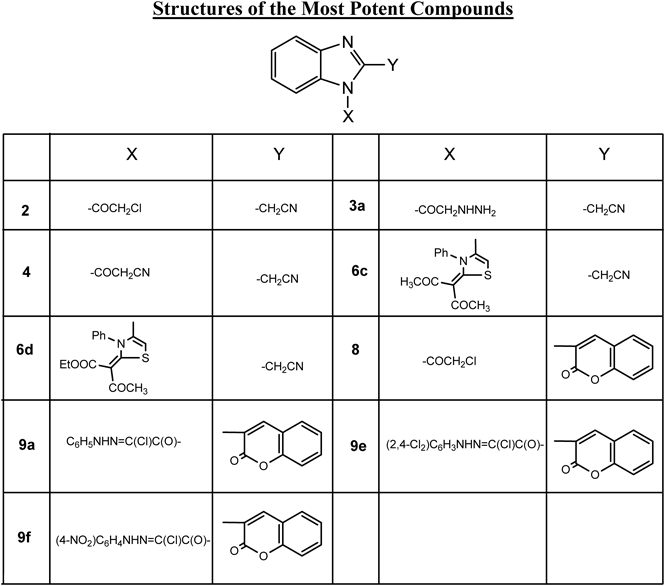
The present work was to discuss a variety and novel benzimidazole derivatives which were synthesized via the reaction of compounds 2 and 8 with different chemical reagents. The results of reactivity of the newly synthesized compounds towards the six cancer cell lines showed that compounds 2 (only for A549 cell line), 3a, 4, 6c, 6d, 8, 9a, 9e and 9f revealed the optimal cytotoxic effect against all the cancer cell lines where foretinib was used as the reference standard. Also, the toxicity of the most cytotoxic nine compounds was evaluated and the result indicated that compounds 3a, 4, 6d, 9e and 9f were non toxic.
All melting points (mp) were uncorrected and determined on an electrothermal apparatus (Büchi 535, Switzerland) in an open capillary tube. IR spectra (KBr discs) were recorded on a FTIR plus 460 IR spectrophotometer (Shimadzu, Japan). 13C-NMR and 1H-NMR spectra were recorded on Varian Gemini-200 (200 MHz) (Japan) spectrometer in DMSO-d6 as solvent, using tetramethylsilane (TMS) as internal reference, and chemical shifts (δ, ppm). Mass spectra were recorded using Hewlett Packard 5988 (U.S.A.). A GC/MS system and GCMS-QP 1000 Ex Shimadzu (Japan) using EI (electron impact method). Elemental analyses were carried out on Vario EL III Elemental CHNS analyzer (Japan).
Synthetic pathways are presented in Charts 1–4, cytotoxicity and toxicity of the newly synthesized products was expressed through Tables 1 and 2.
ChemistryPreparation of the 2-(1H-Benzo[d]imidazol-2-yl)acetonitrile (1)44)To the dry solid of o-phenylene diamine (1.08 g, 0.01 mol), ethyl caynoacetate (1.13 g, 0.01 mol) was added. The reaction mixture was heated in an oil bath at 120°C for 30 min then was left to cool. The solid product produced upon triturating with diethyl ether was collected by filtration and crystallized from ethanol.
Preparation of 2-(1-(2-Chloroacetyl)-1H-benzo[d]imidazol-2-yl)acetonitrile (2)To a compound 1 (1.57 g, 0.01 mol) in 1,4-dioxane (30 mL), chloroacetyl chloride (1.12 g, 0.01 mol) was added. The reaction mixture was heated under reflux for 15 min then poured into a beaker containing ice/water mixture. The formed solid product was collected by filtration, dried and crystallized from 1,4-dioxane.
Pale yellow crystals; mp:>300°C; IR (KBr, cm−1): 3080, 2962, 2196, 1591, 1470. 1H-NMR (DMSO-d6) δ: 3.55 (s, 2H, CH2), 4.38 (s, 2H, CH2), 7.25–7.54 (m, 4H, C6H4). 13C-NMR (DMSO-d6) δ: 14.5, 46.1, 119.5, 112.1, 113.0, 123.5, 123.7, 130.4, 138.0, 164.1, 174.0. MS (EI): m/z (%) 235 [M+2]+ (10.13), 234 [M+1]+ (4.85), 233 [M]+ (30.17), 232 [M−1]+ (0.88), 231 [M−2]+ (0.34), 184 (100.00). Anal. Calcd. for C11H8N3OCl (233.65): C, 56.54; H, 3.45; N, 17.98. Found: C, 56.69; H, 3.70; N, 18.30.
General Procedure for the Preparation of 2-(1-(2-Hydrazinylacetyl)-1H-benzo[d]imidazol-2-yl)acetonitrile Derivatives (3a, b)To a solution of compound 2 (2.33 g, 0.01 mol) in 1,4-dioxane (40 mL), either of hydrazine hydrate (0.50 g, 0.01 mol) or phenyl hydrazine (1.08 g, 0.01 mol) was added. The reaction mixture, in each case, was heated under reflux for 5 h and then poured onto a beaker containing ice/water mixture. The formed solid product, in each case, was collected by filtration, dried and crystallized from 1,4-dioxane.
2-(1-(2-Hydrazinylacetyl)-1H-benzo[d]imidazol-2-yl)acetonitrile (3a)Brown crystals; mp: 273–275°C; IR (KBr, cm−1): 3393, 3229, 3090, 2933, 2192, 1590, 1469. 1H-NMR (DMSO-d6) δ: 2.73 (s, 2H, NH2), 3.90 (s, 2H, CH2), 4.45 (s, 2H, CH2), 7.07–7.67 (m, 4H, C6H4), 12.95 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 16.2, 52.3, 114.7, 115.0, 117.3, 123.4, 123.4, 130.2, 139.1, 167.1, 177.2. MS (EI): m/z (%) 229 [M]+ (11.21), 228 [M−1]+ (48.07), 227 [M−2]+ (12.31), 157 (100.00). Anal. Calcd for C11H11N5O (229.24): C, 57.63; H, 4.84; N, 30.55. Found: C, 58.01; H, 4.99; N, 30.80.
2-(1-(2-(2-Phenylhydrazinyl)acetyl)-1H-benzo[d]imidazol-2-yl)acetonitrile (3b)Brown crystals; mp: 183–185°C; IR (KBr, cm−1): 3427, 3200, 3060, 2928, 2197, 1597, 1446. 1H-NMR (DMSO-d6) δ: 3.40 (s, 2H, CH2), 3.55 (s, 2H, CH2), 6.99–7.94 (m, 9H, C6H4, C6H5), 12.70, 13.30 (2s, 2H, 2NH). 13C-NMR (DMSO-d6) δ: 16.1, 52.1, 114.4, 114.4, 114.8, 115.5, 117.1, 122.3, 123.0, 123.2, 129.3, 129.3, 130.1, 139.0, 141.1, 168.2, 178.1. MS (EI): m/z (%) 307 [M+2]+ (1.40), 306 [M+1]+ (1.19), 305 [M]+ (2.27), 304 [M−1]+ (0.85), 303 [M−2]+ (1.91). Anal. Calcd for C17H15N5O (305.33): C, 66.87; H, 4.95; N, 22.94. Found: C, 66.99; H, 5.20; N, 23.11.
Preparation of 3-(2-(Cyanomethyl)-1H-benzo[d]imidazol-1-yl)-3-oxopropanenitrile (4)A solution of compound 2 (2.33 g, 0.01 mol) in 1,4-dioxane (25 mL) was heated on a water bath at 60°C then potassium cyanide (0.65 g, 0.01 mol) in a least amount of water, was added while stirring. The whole reaction mixture was heated in a water bath at 60°C for 30 min then poured into a beaker containing ice/water mixture with a few drops of hydrochloric acid. The solid product formed was collected by filtration, dried and crystallized from ethanol.
White crystals; mp: 298–300°C; IR (KBr, cm−1): 3009, 2952, 2886, 2210, 2197, 1622, 1593, 1467. 1H-NMR (DMSO-d6) δ: 3.59 (s, 2H, CH2), 4.38 (s, 2H, CH2), 7.26–7.55 (m, 4H, C6H4). 13C-NMR (DMSO-d6) δ: 16.0, 19.1, 112.1, 113.1, 116.1, 117.0, 123.4, 124.0, 130.3, 139.0, 162.2, 174.1. MS (EI): m/z (%) 226 [M+2]+ (0.67), 225 [M+1]+ (1.21), 224 [M]+ (5.51), 223 [M−1]+ (0.67), 57 (100.00). Anal. Calcd for C12H8N4O (224.22): C, 64.28; H, 3.60; N, 24.99. Found: C, 64.67; H, 3.93; N, 25.30.
Preparation of 2-((2-(2-(Cyanomethyl)-1H-benzo[d]imidazol-1-yl)-2-oxoethyl)-amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (5)To a solution of compound 2 (2.33 g, 0.01 mol) in 1,4-dioxane (25 mL), potassium carbonate (0.50 g) and 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (1.78 g, 0.01 mol) were added. The reaction mixture was heated in a boiling water bath for 5 h then poured onto ice/water mixture containing few drops of hydrochloric acid. The solid product formed, was collected by filtration, dried and crystallized from 1,4-dioxane.
Faint brown crystals; mp: 259–261°C; IR (KBr, cm−1): 3445, 3327, 2926, 2841, 2205, 2194, 1617, 1585, 1470. 1H-NMR (DMSO-d6) δ: 1.69–1.71 (m, 4H, 2CH2 cyclohexene), 2.49–2.50 (m, 4H, 2CH2 cyclohexene), 3.56 (s, 2H, CH2), 4.54 (s, 2H, CH2), 6.85–7.64 (m, 4H, C6H4), 12.96 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 16.0, 21.7, 22.8, 23.4, 23.9, 51.9, 83.3, 114.6, 115.9, 116.8, 118.9, 123.4, 123.7, 130.3, 134.5, 139.0, 150.6, 160.0, 162.6, 172.1. MS (EI): m/z (%) 375 [M]+ (59.77), 374 [M−1]+ (72.41), 314 (100.00). Anal. Calcd for C20H17N5OS (375.45): C, 63.98; H, 4.56; N, 18.65; S, 8.54. Found: C, 64.20; H, 4.90; N, 18.90; S, 8.91.
General Procedure for the Preparation of 2-(1-(2-Methylene-3-phenyl-2,3-dihydrothiazol-4-yl)-1H-benzo[d]imidazol-2-yl)acetonitrile (6a–d)To a solution of any of malononitrile (0.66 g, 0.01 mol), ethyl cyanoacetate (1.13 g, 0.01 mol), acetyl acetone (1.00 g, 0.01 mol) or ethyl acetoacetate (1.30 g, 0.01 mol) in dimethylformamide (20 mL), phenyl isothiocyanate (1.35 g, 0.01 mol) containing potassium hydroxide (0.56 g, 0.01 mol) was added, with continuous stirring overnight at room temperature. In the second day, compound 2 (2.33 g, 0.01 mol) was added to each reaction mixture with stirring for another night at room temperature. The whole reaction mixture, in each case, was poured onto ice/water mixture containing a few drops of hydrochloric acid. The solid product formed, in each case, was collected by filtration, dried and crystallized from1,4-dioxane.
2-(4-(2-(Cyanomethyl)-1H-benzo[d]imidazol-1-yl)-3-phenylthiazol-2(3H)-ylidene)malononitrile (6a)Faint brown crystals; mp: 205–207°C; IR (KBr, cm−1): 3100, 3070, 2924, 2260, 2202, 2170, 1627, 1435. 1H-NMR (DMSO-d6) δ: 4.35 (s, 2H, CH2), 6.40 (s, 1H, CH thiazole), 7.15–7.63 (m, 9H, C6H4, C6H5). 13C-NMR (DMSO-d6) δ: 16.0, 50.1, 82.0, 114.0, 115.2, 118.9, 118.9, 119.9, 122.3, 122.6, 122.6, 123.2, 123.6, 129.5, 129.5, 131.0, 133.3, 139.0, 141.1, 165.1, 174.7. MS (EI): m/z (%) 382 [M+2]+ (1.52), 381 [M+1]+ (2.48), 380 [M]+ (0.82), 379 [M−1]+ (1.05), 378 [M−2]+ (0.49), 57 (100.00). Anal. Calcd for C21H12N6S (380.43): C, 66.30; H, 3.18; N, 22.09; S, 8.43. Found: C, 66.60; H, 3.40; N, 22.30; S, 8.80.
Ethyl 2-Cyano-2-(4-(2-(cyanomethyl)-1H-benzo[d]imidazol-1-yl)-3-phenyl-thiazol-2(3H)-ylidene)acetate (6b)Faint brown crystals; mp: 218–220°C; IR (KBr, cm−1): 3062, 2979, 2928, 2201, 2190, 1751, 1623, 1470. 1H-NMR (DMSO-d6) δ: 1.16–1.21 (t, 3H, CH3), 4.14–4.21 (q, 2H, CH2), 4.38 (s, 2H, CH2), 7.38 (s, 1H, CH thiazole), 7.40–7.53 (m, 9H, C6H4, C6H5). 13C-NMR (DMSO-d6) δ: 14.0, 16.6, 60.9, 82.5, 103.6, 114.2, 115.3, 117.0, 117.8, 122.3, 122.7, 122.7, 125.3, 125.3, 129.1, 129.1, 130.3, 134.7, 138.5, 141.1, 164.9, 172.2, 173.3. MS (EI): m/z (%) 427 [M]+ (15.33), 426 [M−1]+ (15.33), 425 [M−2]+ (19.81), 57 (100.00). Anal. Calcd for C23H17N5O2S (427.48): C, 64.62; H, 4.01; N, 16.38; S, 7.50. Found: C, 64.99; H, 4.40; N, 16.60; S, 7.80.
2-(1-(2-(2,4-Dioxopentan-3-ylidene)-3-phenyl-2,3-dihydrothiazol-4-yl)-1H-benzo[d]imidazol-2-yl)acetonitrile (6c)Yellow crystals; mp: 228–230°C; IR (KBr, cm−1): 3070, 2925, 2192, 1651, 1645, 1587, 1460. 1H-NMR (DMSO-d6) δ: 2.49–2.51 (s, 3H, CH3), 2.55–2.57 (s, 3H, CH3), 4.39 (s, 2H, CH2), 7.15 (s, 1H, CH thiazole), 7.27–7.57 (m, 9H, C6H4, C6H5). 13C-NMR (DMSO-d6) δ: 17.7, 30.8, 30.8, 82.3, 112.1, 114.2, 115.3, 119.3, 121.0, 122.8, 122.8, 123.3, 123.3, 129.6, 129.6, 130.4, 136.1, 138.0, 140.2, 163.1, 179.4, 179.4, 183.2. MS (EI): m/z (%) 416 [M+2]+ (3.10), 415 [M+1]+ (11.25), 414 [M]+ (37.64), 413 [M−1]+ (3.98), 412 [M−2]+ (0.97), 77 [C6H5]+ (84.23). Anal. Calcd for C23H18N4O2S (414.48): C, 66.65; H, 4.38; N, 13.52; S, 7.74. Found: C, 66.98; H, 4.63; N, 13.80; S, 8.10.
Ethyl 2-(4-(2-(Cyanomethyl)-1H-benzo[d]imidazol-1-yl)-3-phenylthiazol-2(3H)-ylidene)-3-oxobutanoate (6d)Faint brown crystals; mp: 267–269°C; IR (KBr, cm−1): 3006, 2884, 2195, 1719, 1622, 1590, 1472. 1H-NMR (DMSO-d6) δ: 1.30–1.35 (t, 3H, CH3), 2.03 (s, 3H, CH3), 4.16–4.21 (q, 2H, CH2), 4.39 (s, 2H, CH2), 6.90 (s, 1H, CH thiazole), 7.26–7.55 (m, 9H, C6H4, C6H5). 13C-NMR (DMSO-d6) δ: 14.1, 16.1, 32.7, 60.6, 82.3, 105.2, 112.0, 113.1, 119.4, 122.1, 123.4, 123.4, 124.0, 124.0, 129.5, 129.5, 130.3, 137.2, 138.2, 141.0, 150.5, 164.0, 165.2, 183.1. MS (EI): m/z (%) 445 [M+1]+ (0.68), 444 [M]+ (0.86), 184 (100.00), 77 [C6H5]+ (31.32). Anal. Calcd for C24H20N4O3S (444.51): C, 64.85; H, 4.54; N, 12.60; S, 7.21. Found: C, 65.10; H, 4.88; N, 12.90; S, 7.50.
Preparation of 3-(1H-Benzo[d]imidazol-2-yl)-2H-chromen-2-one (7)To a solution of compound 1 (1.57 g, 0.01 mol) in 1,4-dioxane (30 mL) containing piperidine (0.50 mL), salicylaldehyde (1.22 g, 0.01 mol) was added. The reaction mixture was heated under reflux for 5 h then poured onto ice/water mixture containing a few drops of hydrochloric acid. The formed solid product was collected by filtration, dried and crystallized from1,4-dioxane.
Canary yellow crystals; mp: 228–230°C; IR (KBr, cm−1): 3341, 3051, 2929, 1707, 1602, 1451. 1H-NMR (DMSO-d6) δ: 6.77–8.00 (m, 8H, C6H4), 8.59 (s, 1H, pyran C4), 12.93 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 116.1, 116.6, 116.6, 122.1, 122.7, 122.7, 125.0, 127.1, 129.4, 129.5, 134.8, 134.8, 142.2, 145.7, 153.2, 159.2. MS (EI): m/z (%) 261 [M−1]+ (37.1), 244 (100.00). Anal. Calcd for C16H10N2O2 (262.26): C, 73.27; H, 3.84; N, 10.68. Found: C, 73.50; H, 4.02; N, 10.40.
Preparation of 3-(1-(2-Chloroacetyl)-1H-benzo[d]imidazol-2-yl)-2H-chromen-2-one (8)To a solution of compound 7 (2.62 g, 0.01 mol) in 1,4-dioxane (30 mL), chloroacetyl chloride (1.12 g, 0.01 mol) was added. The reaction mixture was heated under reflux for about 30 min. and then poured onto ice/water mixture. The solid product formed was collected by filtration, dried and crystallized from 1,4-dioxane.
Cannary yellow crystals; mp: 284–286°C; IR (KBr, cm−1): 3058, 2949, 1729, 1609, 1448. 1H-NMR (DMSO-d6) δ: 4.36 (s, 2H, CH2), 7.22–7.73 (m, 8H, 2C6H4), 8.00–8.03 (s, 1H, pyran C4). 13C-NMR (DMSO-d6) δ: 46.0, 114.5, 115.0, 116.6, 120.4, 123.1, 123.3, 125.7, 127.0, 128.0, 130.0, 132.5, 135.0, 146.5, 153.7, 162.0, 169.1, 175.0. MS (EI): m/z (%) 336 [M−2]+ (18.8), 173 (100.00), 77 [C6H5]+ (31.32). Anal. Calcd for C18H11N2O3Cl (338.74): C, 63.82; H, 3.27; N, 8.27. Found: C, 64.10; H, 3.50; N, 8.50.
General Procedure for the Preparation of 2-Oxo-2-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)-N′-phenylacetohydrazonoyl Chloride Derivatives (9a–f)To a cold solution (0–5°C) of 8 (3.38 g, 0.01 mol) in ethanol (50 mL) containing sodium hydroxide solution (10 mL, 10%) a solution (0.01 mol) of any of benzenediazonium chloride, p-chlorobenzenediazonium chloride, p-methoxybenzenediazonium chloride, p-methylbenzenediazonium chloride, 2,4-dichlorobenzenediazonium chloride or p-nitrobenzenediazonium chloride [which was prepared by the addition of sodium nitrite (0.69 g, 0.01 mol) in water (2 mL) to a cold solution (0–5°C) of the corresponding arylamine (0.01 mol) containing the appropriate amount of hydrochloric acid (3 mL) with continuous stirring] was added with continuous stirring. The reaction mixture was stirred at room temperature and the solid product formed was collected by filtration and dried. The obtained product was crystallized from ethanol.
2-Oxo-2-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)-N′-phenylacetohydrazonoyl Chloride (9a)Dark yellow crystals; mp: 213–215°C; IR (KBr, cm−1): 3410, 3053, 1711, 1650, 1606, 1567, 1510, 1445. 1H-NMR (DMSO-d6) δ: 7.21–7.91 (m, 13H, 2C6H4, C6H5), 8.11 (s, 1H, pyran C4), 9.25 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 113.0, 113.0, 114.0, 115.0, 120.5, 122.6, 123.0, 123.4, 124.8, 125.4, 126.8, 128.0, 129.5, 129.6, 129.6, 130.5, 134.5, 142.0, 142.4, 145.6, 162.2, 168.1, 175.0, 179.3. MS (EI): m/z (%) 445 [M+2]+ (0.68), 444 [M+1]+ (1.92), 443 [M]+ (4.04), 442 [M−1]+ (11.04), 441 [M−2]+ (4.78), 262 (100.00), 77 [C6H5]+ (86.99). Anal. Calcd for C24H15N4O3Cl (442.85): C, 65.09; H, 3.41; N, 12.65. Found: C, 65.30; H, 3.71; N, 12.99.
N′-(4-Chlorophenyl)-2-oxo-2-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)acetohydrazonoyl Chloride (9b)Pale orange crystals; mp: 280–282°C; IR (KBr, cm−1): 3410, 3302, 3035, 1709, 1606, 1577, 1511, 1475. 1H-NMR (DMSO-d6) δ: 7.24–7.93 (m, 12H, 3C6H4), 8.13 (s, 1H, pyran C4), 9.26 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 114.3, 115.3, 116.2, 117.4, 117.4, 119.1, 123.2, 123.2, 125.2, 127.6, 127.8, 128.4, 129.4, 129.4, 129.5, 130.3, 137.1, 142.7, 148.2, 150.0, 155.1, 161.5, 167.2, 175.1. MS (EI): m/z (%) 478 [M+1]+ (0.11), 477 [M]+ (0.15), 476 [M−1]+ (0.15), 475 [M−2]+ (0.26), 261 (100.00). Anal. Calcd for C24H14N4O3Cl2 (477.30): C, 60.39; H, 2.96; N, 11.74. Found: C, 60.69; H, 3.31; N, 11.90.
N′-(4-Methoxyphenyl)-2-oxo-2-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)acetohydrazonoyl Chloride (9c)Brown crystals; mp: 213–215°C; IR (KBr, cm−1): 3335, 3056, 2928, 1707, 1601, 1570, 1499, 1452. 1H-NMR (DMSO-d6) δ: 3.87 (s, 3H, CH3), 7.13–8.01 (m, 12H, 3C6H4), 8.10 (s, 1H, pyran C4), 9.14 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 55.6, 114.4, 115.2, 115.5, 115.5, 116.0, 117.2, 117.2, 119.4, 123.9, 123.9, 124.9, 127.8, 128.3, 130.2, 129.5, 132.9, 138.5, 145.5, 153.2, 154.2, 158.7, 161.7, 167.5, 175.3. MS (EI): m/z (%) 475 [M+2]+ (0.28), 474 [M+1]+ (0.70), 473 [M]+ (0.70), 472 [M−1]+ (0.14), 471 [M−2]+ (0.08), 396 (100.00). Anal. Calcd for C25H17N4O4Cl (472.88): C, 63.50; H, 3.62; N, 11.85. Found: C, 63.81; H, 3.91; N, 12.10.
2-Oxo-2-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)-N′-(p-tolyl)acetohydrazonoyl Chloride (9d)Orange crystals; mp: 223–225°C; IR (KBr, cm−1): 3394, 3053, 2922, 1709, 1603, 1570, 1508, 1453. 1H-NMR (DMSO-d6) δ: 2.40 (s, 3H, CH3), 7.21–8.01 (m, 12H, 3C6H4), 8.14 (s, 1H, pyran C4), 9.16 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 55.5, 114.5, 115.3, 115.9, 115.9, 116.5, 117.3, 117.3, 119.4, 122.5, 122.5, 125.0, 127.8, 128.5, 129.4, 130.3, 132.8, 138.2, 145.6, 153.1, 154.4, 158.7, 161.5, 167.3, 176.2. MS (EI): m/z (%) 456 [M−1]+ (0.29), 455 [M−2]+ (0.78), 91 (100.00), 77 [C6H5]+ (12.31). Anal. Calcd for C25H17N4O3Cl (456.88): C, 65.72; H, 3.75; N, 12.26. Found: C, 65.99; H, 4.01; N, 12.30.
N′-(2,4-Dichlorophenyl)-2-oxo-2-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)acetohydrazonoyl Chloride (9e)Faint brown crystals; mp: 278–280°C; IR (KBr, cm−1): 3385, 3059, 1706, 1600, 1573, 1510, 1457. 1H-NMR (DMSO-d6) δ: 7.14–8.07 (m, 11H, 2C6H4, C6H3), 8.14 (s, 1H, pyran C4), 9.20 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 113.3, 114.6, 118.0, 118.4, 120.4, 123.2, 123.2, 125.2, 125.8, 126.1, 126.7, 127.2, 128.2, 130.1, 132.8, 135.6, 137.1, 145.5, 146.0, 155.3, 161.5, 167.3, 170.3, 172.2. MS (EI): m/z (%) 511 [M]+ (0.14), 510 [M−1]+ (0.29), 509 [M−2]+ (0.84), 261 (100.00). Anal. Calcd for C24H13N4O3Cl3 (511.74): C, 56.33; H, 2.56; N, 10.95. Found: C, 56.69; H, 2.82; N, 11.30.
N′-(4-Nitrophenyl)-2-oxo-2-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)acetohydrazonoyl Chloride (9f)Brown crystals; mp: 243–245°C; IR (KBr, cm−1): 3408, 3060, 1716, 1601, 1570, 1519, 1456. 1H-NMR (DMSO-d6) δ: 7.21–7.98 (m, 12H, 3C6H4), 8.17 (s, 1H, pyran C4), 9.17 (s, 1H, NH). 13C-NMR (DMSO-d6) δ: 113.5, 113.5, 114.2, 115.6, 116.6, 120.1, 123.2, 123.2, 124.0, 124.0, 125.2, 127.0, 127.3, 130.0, 131.3, 137.3, 138.6, 145.7, 148.9, 153.7, 161.5, 167.1, 170.3, 173.5. MS (EI): m/z (%) 490 [M++2] (0.03), 489 [M+1]+ (0.04), 488 [M]+ (0.04), 487 [M−1]+ (0.03), 486 [M−2]+ (0.08), 261 (100.00). Anal. Calcd for C24H14N5O5Cl (487.85): C, 59.09; H, 2.89; N, 14.36. Found: C, 59.29; H, 3.11; N, 14.60.
Preparation of 3-Oxo-3-(2-(2-oxo-2H-chromen-3-yl)-1H-benzo[d]imidazol-1-yl)propanenitrile (10)A solution of compound 8 (3.38 g, 0.01 mol) in 1,4-dioxane (25 mL) was heated on water bath at 60°C, then potassium cyanide (0.65 g, 0.01 mol) in a least amount of water (2 mL), was added with continuous stirring. The reaction mixture was left in the water bath for 30 min. at 60°C then poured onto ice/water mixture containing a few drops of hydrochloric acid. The solid product, so formed, was collected by filtration and crystallized from ethanol.
Brown crystals; mp: 210–212°C; IR (KBr, cm−1): 3065, 2930, 2201, 1714, 1647, 1610, 1457. 1H-NMR (DMSO-d6) δ: 3.56 (s, 2H, CH2), 7.12–7.18 (s, 1H, pyran C4), 7.20–7.95 (m, 8H, 2C6H4). 13C-NMR (DMSO-d6) δ: 113.5, 114.2, 115.1, 116.7, 120.7, 123.8, 123.8, 125.0, 126.2, 127.5, 130.5, 133.5, 138.5, 142.1, 150.2, 161.2, 165.8, 172.3. MS (EI): m/z (%) 331 [M+2]+ (0.19), 330 [M+1]+ (0.25), 329 [M]+ (0.37), 327 [M−2]+ (0.25), 57 (100.00). Anal. Calcd for C19H11N3O3 (329.31): C, 69.30; H, 3.37; N, 12.76. Found: C, 69.68; H, 3.71; N, 12.95.
Preparation of 2-((1-(2-Chloroacetyl)-1H-benzo[d]imidazol-2-yl)methyl)thiazol-4(5H)-one (11)To a solution of compound 2 (2.33 g, 0.01 mol) in acetic acid (15 mL), thioglycollic acid (0.92 g, 0.01 mol) was added. The reaction mixture was heated under reflux system for about 5 h and then poured onto ice/water mixture. The solid product formed was collected by filtration, dried and crystallized from 1,4-dioxane.
Pale yellow crystals; mp: 295–297°C; IR (KBr, cm−1): 3082, 3010, 2955, 2885, 1713, 1624, 1590, 1469. 1H-NMR (DMSO-d6) δ: 3.38 (s, 2H, CH2), 3.70 (s, 2H, CH2), 4.38 (s, 2H, CH2), 7.26–7.55 (m, 4H, C6H4). 13C-NMR (DMSO-d6) δ: 38.7, 39.8, 45.8, 112.1, 113.0, 123.1, 123.5, 130.4, 139.0, 164.1, 168.0, 172.3, 178.1. MS (EI): m/z (%) 310 [M+2]+ (5.61), 309 [M+1]+ (4.43), 57 (100.00). Anal. Calcd for C13H10N3O2SCl (307.76): C, 50.73; H, 3.28; N, 13.65; S, 10.42. Found: C, 50.99; H, 3.51; N, 13.90; S, 10.66.
General Procedure for the Preparation of 5-Amino-2-((1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)methyl)-7-phenyl-7H-pyrano[2,3-d]thiazole-6-carbonitrile Derivatives (12a–d)To a solution of compound 11 (3.07 g, 0.01 mol) in 1,4-dioxane (20 mL) containing a catalytic amount of triethylamine (0.50 mL) any of benzaldehyde (1.06 g, 0.01 mol), p-chlorobenzaldehyde (1.40 g, 0.01 mol), p-methoxybenzaldehyde (1.36 g, 0.01 mol) or 2-nitrobenzaldehyde (1.51 g, 0.01 mol) and malononitrile (0.66 g, 0.01 mol) were added. The reaction mixture, in each case, was heated under reflux system for 5 h then poured onto ice/water mixture containing a few drops of hydrochloric acid. The solid products formed, in each case, was collected by filtration, dried and crystallized from1,4-dioxane.
5-Amino-2-((1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)methyl)-7-phenyl-7H-pyrano[2,3-d]thiazole-6-carbonitrile (12a)Yellow crystals; mp: 290–292°C; IR (KBr, cm−1): 3428, 3231, 3077, 2953, 2884, 2198, 1621, 1589, 1470. 1H-NMR (DMSO-d6) δ: 3.56 (s, 2H, CH2), 4.38 (s, 2H, CH2), 4.60 (s, 1H, pyran C4), 7.23–7.93 (m, 9H, C6H4, C6H5), 8.53 (s, 2H, NH2). 13C-NMR (DMSO-d6) δ: 29.0, 30.0, 45.8, 59.0, 114.1, 115.0, 119.5, 123.1, 123.5, 125.0, 128.0, 128.0, 129.4, 129.4, 130.4, 134.3, 139.0, 141.0, 144.0, 150.6, 160.2, 164.0, 183.2. MS (EI): m/z (%) 462 [M]+ (0.30), 461 [M−1]+ (0.31), 233 (100.00). Anal. Calcd for C23H16N5O2SCl (461.92): C, 59.80; H, 3.49; N, 15.16; S, 6.94. Found: C, 59.91; H, 3.81; N, 15.30; S, 7.20.
5-Amino-2-((1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)methyl)-7-(4-chlorophenyl)-7H-pyrano[2,3-d]thiazole-6-carbonitrile (12b)Brown crystals; mp: 264–266°C; IR (KBr, cm−1): 3429, 3226, 3097, 2952, 2884, 2196, 1621, 1587, 1470. 1H-NMR (DMSO-d6) δ: 3.57 (s, 2H, CH2), 4.38 (s, 2H, CH2), 4.40 (s, 1H, pyran C4), 7.26–7.97 (m, 8H, 2C6H4), 8.54 (s, 2H, NH2). 13C-NMR (DMSO-d6) δ: 29.8, 30.5, 45.8, 64.0, 114.2, 115.5, 119.4, 123.4, 123.4, 125.8, 125.8, 129.9, 129.9, 130.2, 131.9, 133.3, 139.0, 141.3, 143.5, 150.4, 160.0, 164.5, 183.0. MS (EI): m/z (%) 498 [M+2]+ (0.13), 497 [M+1]+ (0.42), 496 [M+] (1.98), 495 [M−1]+ (5.83), 494 [M−2]+ (2.02), 57 (100.00). Anal. Calcd for C23H15N5O2SCl2 (496.37): C, 55.65; H, 3.05; N, 14.11; S, 6.46. Found: C, 55.99; H, 3.41; N, 14.30; S, 6.80.
5-Amino-2-((1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)methyl)-7-(4-methoxyphenyl)-7H-pyrano[2,3-d]thiazole-6-carbonitrile (12c)Brown crystals; mp: 271–273°C; IR (KBr, cm−1): 3423, 3224, 3074, 2946, 2196, 1625, 1569, 1467. 1H-NMR (DMSO-d6) δ: 3.55 (s, 3H, CH3), 3.81 (s, 2H, CH2), 4.37 (s, 2H, CH2), 4.90 (s, 1H, pyran C4), 7.11–7.95 (m, 8H, 2C6H4), 8.32 (s, 2H, NH2). 13C-NMR (DMSO-d6) δ: 29.7, 30.5, 45.8, 55.7, 64.0, 113.7, 114.6, 114.6, 115.0, 119.4, 123.3, 123.3, 127.2, 130.2, 130.2, 130.7, 138.4, 141.3, 143.6, 150.4, 157.2, 160.2, 164.2, 183.0. MS (EI): m/z (%) 493 [M++1] (0.18), 492 [M+] (0.18), 184 (100.00). Anal. Calcd for C24H18N5O3SCl (491.95): C, 58.59; H, 3.69; N, 14.24; S, 6.52. Found: C, 58.69; H, 3.82; N, 14.30; S, 6.73.
5-Amino-2-((1-(2-chloroacetyl)-1H-benzo[d]imidazol-2-yl)methyl)-7-(2-nitrophenyl)-7H-pyrano[2,3-d]thiazole-6-carbonitrile (12d)Dark brown crystals; mp: 280–282°C; IR (KBr, cm−1): 3429, 3200, 3100, 2925, 2196, 1624, 1590, 1466. 1H-NMR (DMSO-d6) δ: 3.56 (s, 2H, CH2), 4.38 (s, 2H, CH2), 4.90 (s, 1H, pyran C4), 7.25–7.54 (m, 8H, 2C6H4), 8.01 (s, 2H, NH2). 13C-NMR (DMSO-d6) δ: 25.6, 30.2, 45.8, 64.0, 114.3, 115.2, 119.4, 123.4, 123.4, 124.2, 126.1, 129.7, 130.2, 132.5, 134.1, 138.7, 141.5, 143.2, 149.1, 150.4, 160.7, 164.9, 183.1. MS (EI): m/z (%) 509 [M+2]+ (0.24), 508 [M+1]+ (0.17), 507 [M]+ (0.25), 184 (100.00). Anal. Calcd for C23H15N6O4SCl (506.92): C, 54.49; H, 2.98; N, 16.58; S, 6.33. Found: C, 54.80; H, 3.21; N, 16.90; S, 6.65.
Professor R. M. Mohareb would like to express his deepest thanks to Faculty of Science Cairo University for the financial support through the research project.
The authors declare no conflict of interest.