Review
Studies on Catalytic Activation of Olefins Using Cobalt Complex
2018 Volume 66 Issue 4 Pages 339-346
Details
2018 Volume 66 Issue 4 Pages 339-346
In this review, I tell the story of the cobalt chemistry that has been developed in my group since 2011. First, we achieved the total synthesis of polyketide natural product trichodermatide A, which involved a late-stage Isayama-Mukaiyama hydration of an enol ether using cobalt(II) acetylacetonate (Co(acac)2) that gave the desired product chemo-, regio-, and diastereoselectively. After our report of this total synthesis in 2013, we were required to revise the originally reported structure of trichodermatide A following the accurate and important report from the Trauner group. Second, we found unique cobalt-catalyzed hydroelementation reactions of olefins involving a cobalt-salen complex, N-fluoro-2,4,6-trimethylpyridinium salt, and a silane reagent. Under these reaction conditions, a carbocationic or carbon radical species is generated from an olefin, and then C–X (X=O, N, C, F) bond formation occurs with good functional group tolerance for a broad substrate scope. This review also covers recent examples of switching chemistry and natural product synthesis involving my cobalt chemistry reported by several groups.
Recently, the hydroelementation of unactivated olefins catalyzed by a transition metal such as cobalt, manganese, or iron has been in the spotlight in synthetic organic chemistry and organometallic chemistry.1) In this review, I introduce cobalt chemistry and our achievements in this field.
The milestone development in the history of cobalt-catalyzed hydroelementation is the hydration reaction reported by Isayama and Mukaiyama in 1989.2) They found that an olefin can be hydrated by using cobalt(II) acetylacetonate (Co(acac)2) and PhSiH3 under an oxygen atmosphere in good yield and with perfect Markovnikov selectivity. In my opinion, the value of this chemistry was increased by Carreira’s extensive research on the hydrofunctionalization of olefins.3–5) Carreira’s reactions also required a cobalt catalyst and silane reagent and gave a variety of products depending on the radical trapping reagent. Notably, it is believed that the mechanisms of the original hydration and Carreira’s hydrofunctionalization are similar. Furthermore, around the same time, the cobalt-catalyzed hydration was applied in Kanai and Shibasaki’s synthesis of garsubellin A.6)
Inspired by these impressive works, in 2011, I launched our research on cobalt catalysts for natural product synthesis and the development of new catalytic reactions. In this review, I summarize the achievements in cobalt research. In our cobalt research program, we initially utilized the cobalt-catalyzed hydration to achieve the total synthesis of a complex natural product.
Trichodermatide A (1) was isolated from Trichoderma ressei and its structure was elucidated using NMR spectroscopy and circular dichroism spectral analyses7) (Chart 1). The highly oxygenated pentacyclic structure contains acetal and hemiacetal moieties. From the standpoint of synthesis, the labile hemiacetal must be constructed at a late stage. In this case, the control of stereochemistry is also required.

The synthetic study commenced with the stereocontrolled construction of the tetracyclic framework. Compound 3 was synthesized through diastereoselective acetalization. Diastereoselective hydroxylation of ketone using the Davis reagent gave compound 4. After protection of the hydroxyl group as the 4-nitrobenzoate, diastereoselective allylic oxidation using SeO2 furnished compound 6. Finally, Isayama–Mukaiyama hydration of the enol ether using Co(acac)2 in trifluoroethanol gave compound 7 chemo-, regio-, and diastereoselectively, and this was followed by deprotection to give trichodermatide A (1).8)
In 2014, Trauner and Ochsenfeld reported the synthesis of 2, whose structure was elucidated using X-ray crystallography.9) It is noteworthy that the NMR spectroscopic data of the sample used for the X-ray were not identical to those in the original isolation paper. Carelessly, I had drawn the structure of trichodermatide A as 2 (Chart 1) at the time of the disclosure of our total synthesis in 2013. Because of Trauner’s report, I decided to revisit the trichodermatide A research; our group identified the error in the stereochemistry at C10 in structure 2 through X-ray crystallography and revised the structure to 1 using a synthetic intermediate of a trichodermatide A analog.10)
In parallel with natural product synthesis, I also pushed projects involving the discovery of new cobalt-catalyzed reactions. Our discovery in the field of cobalt research is shown in Chart 2. For the reactions of Isayama, Carreira, and others, it is believed that radical species generated from olefins and cobalt hydride through a hydrogen atom transfer mechanism react with radical trapping reagents to afford products. This type of reaction has been reported extensively. In contrast, our reaction clearly differs in terms of the key reaction intermediate, which is a carbocationic species generated via a radical-cation crossover mechanism. A nucleophile can attack the carbocationic species intermolecularly or intramolecularly to furnish unprecedented products in cobalt hydride chemistry.11,12)

The radical-cation crossover mechanism was found serendipitously during work on our originally planned hydrofluorination (Chart 3). With this research, my goal was to obtain hydrofluorinated products using carbon radicals via the classical mechanism in cobalt hydride chemistry. However, treatment of compound 8 with salen-cobalt complex 11, phenylsilane, and N-fluorobenzenesulfonimide (NFSI) as a radical fluorine source in ethanol gave hydroalkoxylation product 9 in excellent yield instead of hydrofluorination product 10.

The optimal reaction conditions shown in Chart 4 are mild, general, and functional-group tolerant.13,14) In addition to ethanol, other alcoholic solvents such as methanol, bulky tert-butyl alcohol, and weakly nucleophilic trifluoroethanol were also applicable. The reaction mechanism of this catalytic hydroalkoxylation is shown in Chart 5. Cobalt fluoride and cobalt triflate (cationic cobalt species) would be generated from the cobalt complex and N-fluoropyridinium salt. The reaction between cobalt fluoride and the silane reagent would give cobalt hydride, which would react with an olefin to give a carbon radical intermediate. I believe that the radical-cation crossover step occurs owing to the one-electron oxidation of the carbon radical species by remaining cationic cobalt species. Concerted inter- or intramolecular nucleophilic attack would occur to generate a C–Nu bond, as I will discuss later.


In the case of alkenyl alcohols as substrates in toluene, intramolecular hydroalkoxylation produces oxygen-containing heterocycles such as 19–2115) (Chart 6). Analogously, lactones such as 22–24 are formed from alkenyl carboxylic acids.

Catalyst 11 gave unsatisfactory yields for medium-sized rings (more than seven atoms, Chart 7). A thorough investigation revealed that the optimal catalyst, 31, afforded compound 25 in 60% yield whereas complex 11 gave a 30% yield. Furthermore, catalyst 31 gave other medium-sized cyclic ethers and lactones such as 26–30.

These powerful reaction conditions also enable deprotective cyclization of protected substrates (Chart 8). Various protecting groups are applicable. Cyclic ethers can be obtained from tert-butyldimethylsilyl- (TBS), methoxymethyl- (MOM), or Bn-protected alkenyl alcohols. Lactones can be obtained from alkenyl esters; in particular, methyl esters were found to be better than others, such as ethyl, benzyl, and tert-butyl esters.

These reaction conditions also enable C–N bond formation for aminoalkenes (hydroamination of olefins, Chart 9).16) The nitrogen atom must be protected as a sulfonamide or an amide; free amino groups were not applicable. Five-, six-, and seven-membered ring products (36–39) were synthesized. Furthermore, various products were obtained from aminoalkenes that bore, for example, a phenol (40), a cyclopropane (41), and an epoxide (42). Hydroarylation of olefins was realized with alkenyl arenes17) (Chart 10). In the case of 1,1-disubstituted olefins, complex 51 gave the desired products, such as 43–49, with good functional group tolerance. No racemization was observed in product 49. Complex 11 was optimal for the monosubstituted olefin, giving 50.


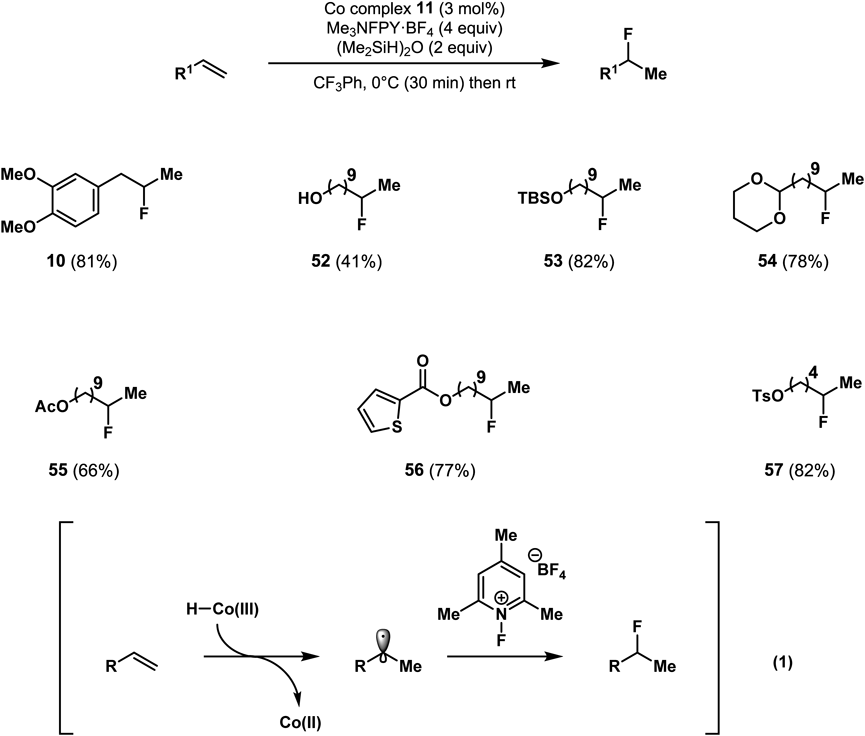
As shown in Chart 3, my original goal was to develop the hydrofluorination of olefins. Based on investigations into hydroalkoxylation research, our group found that hydrofluorination occurs in non-alcoholic solvent trifluorotoluene18) (Chart 11). The proposed mechanism is shown in eq. 1 and involves a direct reaction between a radical species and N-fluoropyridinium tetrafluoroborate. In summary, in the presence of a cobalt-salen complex, an N-fluoropyridinium salt, and a silane reagent, if there is a proper nucleophilic species that can access an olefin intermolecularly or intramolecularly, a radical-cation crossover pathway is created. From the synthesis point of view, I highlight that our method can generate carbocationic species from olefins under neutral conditions and has excellent functional group tolerance and a broad substrate scope.
Finally, I will discuss recent research by other groups in which our cobalt catalyst system was utilized (Chart 12). Diaconescu reported a redox-switchable tetrahedral cobalt complex supported by a ferrocene-based ligand.19) They evaluated the redox-switchability of complex 58 using our hydroalkoxylation. The redox state of complex 58 is active, whereas the oxidized state of complex 59 is inactive. Newhouse and Maimone reported the first total syntheses of andrastin D (64) and terretonin L (66) from a common intermediate (62).20) The framework of andrastin D was constructed using our cobalt catalysis conditions, which generate carbocationic species from olefins under neutral conditions. In contrast, they synthesized intermediate 65 using Carreira’s hydrochlorination21) (TsCl can be replaced with tert-butyl hydroperoxide).

In summary, our group discovered a unique cobalt catalyst system featuring an N-fluoropyridinium salt. Our method is useful in synthetic organic chemistry because of its functional group tolerance. The radical-cation crossover mechanism clearly differentiates our reaction from other cobalt-hydride-related reactions.
The research was supported by JSPS KAKENHI Grant number 26860017, the Uehara memorial foundation, Takeda Science Foundation, the Society of Synthetic Organic Chemistry (Takeda Pharmaceutical Company Award). The author would like to thank Prof. Kou Hiroya, and all the collaborators, friends, and family.
The author declares no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2017 Pharmaceutical Society of Japan Award for Young Scientists.