Regular Articles
Introduction of a Polar Functional Group to the Lipid Tail of 4-epi-Jaspine B Affects Sphingosine Kinase Isoform Selectivity
2018 Volume 66 Issue 9 Pages 866-872
Details
2018 Volume 66 Issue 9 Pages 866-872
Sphingosine kinases (SphKs) are key enzymes that regulate sphingosine 1-phosphate production levels, and are involved in a range of cellular processes. Focusing on a hydrophilic residue in the hydrophobic binding pocket of SphKs, we designed and synthesized 4-epi-jaspine B derivatives containing a polar functional group in the lipid tail. A biological evaluation revealed that the introduction of ether groups to the lipid tail of 4-epi-jaspine B modulated its isoform selectivity toward SphKs.
The sphingosine 1-phosphate (S1P) signaling pathway plays an important role in modulating a range of cellular processes including apoptosis, mitogenesis, lymphocyte trafficking and angiogenesis.1,2) Sphingosine kinases (SphKs) are the key enzymes in the S1P signaling pathway,3–5) which can regulate the production of S1P from sphingosine. There are two isoforms of SphKs in mammals: SphK1 and SphK2, serving the same catalytic reaction, but displaying different cellular localizations. SphK1 exists mainly in the cytosol, and is translocated to the plasma membrane upon phosphorylation at Ser255.6) The elevation of cellular SphK1 levels promotes cell survival, proliferation and migration.5) Therefore, the overexpression of SphK1 is observed in various tumors and is associated with the severity of malignancy. On the other hand, SphK2 functions in various cellular organelles.5) SphK2 in the endoplasmic reticulum and mitochondria is associated with apoptosis. SphK2 in the nucleus is involved in epigenetic regulation. Several studies have revealed that elevated expression of SphK2 is correlated with tumor growth, similar to that of SphK1.7) Furthermore, the roles of SphK2 in autoimmune and inflammatory diseases have been widely investigated using SphK2−/− mice.8) For example, SphK2 regulates interleukin (IL)-12-stimulated interferon gamma production.9) Therefore, SphK2 has recently attracted attention as a therapeutic target for autoimmune and inflammatory disorders.
To date, a number of SphK inhibitors have been developed, including isoform selective inhibitors.3,5) In particular, because of the availability of a crystal structure analysis of SphK1,10,11) the development of potent SphK1 selective inhibitors has been accelerated. In contrast, most of the reported SphK2 inhibitors display moderate potency and selectivity. From a structural point of view, the SphK inhibitors are classified into two groups: nonlipid type inhibitors and lipid (sphingosine) based inhibitors.3) The representative nonlipid type inhibitors, such as SKI-II12) or PF-543,13) are derived from chemical library screening. In contrast, the lipid based inhibitors are sphingosine analogues (DMS,14) K145,15) etc.), which consist of polar head and lipid tail parts. During the course of structure–activity relationship (SAR) studies of lipid based inhibitors, many researchers have focused on the derivatization of polar head groups, which could provide a key interaction with the binding pocket of SphK.3) On the other hand, there is less information about the modification of the lipid tail parts, although several groups have reported that the introduction of an aromatic residue16) or bulky substituents such as cyclohexane17) to the lipid tail affected its inhibitory potency.
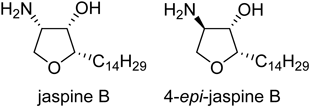
Our group has focused on a naturally occurring sphingosine analogue, jaspine B18,19) (Fig. 1), as a promising lead for the development of novel SphK inhibitors. Through synthetic20–23) and SAR studies,24–26) a number of jaspine B derivatives with modification at the polar head group and lipid tail have been reported. For example, our examination of modifying the polar head has revealed that a 4-epi isomer of jaspine B (4-epi-jaspine B) displays the most potent inhibitory activity among the eight possible stereoisomers24) (Fig. 1). The introduction of a phenylene tether to the lipid tail enhanced SphK2 selectivity, and the attachment of cyclohexyl groups to the lipid terminal parts resulted in a slight improvement in the inhibitory activity of both SphK isoforms.25) We also reported that the replacement of a methylene tether with an oxygen atom (ether group) altered the potency of the 4-epi-jaspine B.25) These results prompted us to systematically investigate the introduction of polar functional group(s) to the lipid tail of jaspine B analogues. In the present study, we have focused on an SAR at the lipid tail for inhibitory activities toward SphKs.
To undertake this SAR study, we paid our attention to a water-shielded hydrogen bond between SphKs and jaspine B derivatives. The water-shielded hydrogen bond represents a characteristic hydrogen bond, which is located deep inside the binding pocket.27–29) When the water accessibility to a buried polar group in the binding sites is limited, the hydrogen bond(s) between the protein and ligand cannot be easily dissociated because of the unlikely simultaneous hydration by solvent water molecule(s). Consequently, the tight and long-lived protein–ligand complexes can be formed. Barril et al. reported that the kinetic stability of the receptor-ligand complexes was enhanced by formation of a shielded hydrogen bond at a deep site in the binding pocket of the receptor.29) Additionally, several studies have shown that the hydrophobicity of the surrounding environments in the binding pocket affected the hydrogen bond strengths.30,31) Our group has recently developed two types of novel immunomodulators: glycolipid CD1d ligands32) and lipopeptide Toll-like receptor 2 (TLR2) ligands33) containing a polar functional group in their lipid tail. In these investigations, the binding affinities and immunological activities of these ligands varied depending on the position of the polar functional group, which would be attributed to the formation of shielded hydrogen bonds in the hydrophobic lipid binding pockets.
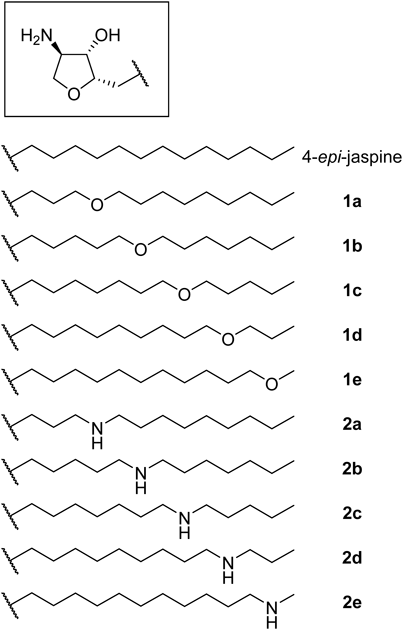
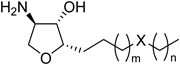
According to the crystal structure analysis of SphK1, the lipid tail of sphingosine is accommodated in a buried hydrophobic pocket surrounded by hydrophobic residues.10) We focused on a hydrophilic threonine (Thr) residue (Thr196) in the hydrophobic pocket (Fig. 2a), which could contribute to anchoring the potential ligands via water-shielded hydrogen bond(s). The crystal structure analysis of SphK1 complexed with an inhibitor SKI-II also revealed that an amino group of SKI-II was within hydrogen bond distance to the β-hydroxy group of Thr19610) (Fig. 2b). SphK2 also contains the corresponding hydrophilic amino acid residue (Thr326) at a similar position.10) On the basis of these information, we designed a series of 4-epi-jaspine B derivatives containing two polar functional groups in their lipid tail: (1) ether group (–O–) in the lipid tail (1a–e) and (2) amino group (–NH–) in the lipid tail (2a–e) (Fig. 3).

The synthesis of the designed derivatives containing polar functional groups (ether or amino groups) is shown in Chart 1. We employed a previously reported synthesis based on a late stage cross metathesis reaction that facilitates the introduction of various lipid side chains to the tetrahydrofuran head group.24) The benzyloxycarbonyl (Cbz)-protected amine 4 was prepared from tetrahydrofuran 323) by removal of the tert-butoxycarbonyl (Boc) group with 4M HCl in dioxane followed by protection using Cbz-Cl in 36% yield. We next modified the tetrahydrofuran head group with a variety of lipid tails. The cross metathesis reaction of 4 with lipid side chain synthons 5a–e or 6a–e in the presence of Hoveyda–Grubbs II catalyst provided the unsaturated precursor, which was treated using Pd(OH)2/C under hydrogen atmosphere to lead to desired derivatives 1a–e, 2a–e.

With a series of 4-epi-jaspine B derivatives in hand, we next evaluated their in vitro inhibitory activities toward SphK1 and SphK2, using an off-chip mobility shift assay (Table 1). We employed 4-epi-jaspine B as a reference SphK inhibitor. The 5′-, 11′- and 13′-oxo derivatives 1a, 1d and 1e displayed weaker activities toward SphK1, compared with 4-epi-jaspine B, while the 7′- and 9′-oxo derivatives 1b and 1c exhibited moderate inhibitory activities toward SphK1 (IC50=15 and 22 µM). The arrangement of an ether oxygen at the middle part in the lipid tail was relatively favorable for the inhibitory activities, implying that site-specific interaction between a hydrogen acceptor of ether oxygen and the polar residues in SphK1 might affect the ligand binding interactions. Interestingly, most of the derivatives with ether groups retained their inhibitory activities toward SphK2 (IC50=2.7–9.1 µM) with the exception of 5′-oxo derivative 1a (IC50=21 µM). In terms of isoform selectivity, particularly, the 11′-oxo derivative 1d effectively inhibited SphK2 (IC50=3.2 µM) with no inhibition of SphK1, even at 30 µM.34) The SphK1 and SphK2 inhibitory activities of the aza-derivative (2a–e) were markedly reduced compared with those of 4-epi-jaspine B and the derivatives with an ether group.
 | |||||
|---|---|---|---|---|---|
| Compound | X | m | n | IC50 (µM)a) | |
| SphK1 | SphK2 | ||||
| 4-epi-Jaspine B | 5.3 | 1.6 | |||
| 1a | O | 2 | 8 | >30 | 21 |
| 1b | O | 4 | 6 | 15 | 2.7 |
| 1c | O | 6 | 4 | 22 | 5.6 |
| 1d | O | 8 | 2 | >30 | 3.2 |
| 1e | O | 10 | 0 | >30 | 9.1 |
| 2a | NH | 2 | 8 | >30 | >30 |
| 2b | NH | 4 | 6 | >30 | >30 |
| 2c | NH | 6 | 4 | >30 | >30 |
| 2d | NH | 8 | 2 | >30 | >30 |
| 2e | NH | 10 | 0 | >30 | >30 |
a) IC50 values are the concentration required for 50% inhibition of the sphingosine phosphorylation by SphK1 or SphK2.
In conclusion, we designed and synthesized 4-epi-jaspine B derivatives with an ether or an amino group in the lipid tail. The derivatives with amino groups displayed no inhibitory activities toward both SphK isoforms. In contrast, some of the derivatives with ether groups moderately inhibited the activity of SphK1, and most of the ether group-containing derivatives preserved the activity of 4-epi-jaspine B toward SphK2. Intriguingly, replacement of one methylene at the lipid tail with ether oxygen affected the inhibitory activity of 4-epi-jaspine B, leading to the identification of a selective SphK2 inhibitor, such as the 11′-oxo derivative 1d [SphK1 (IC50 >30 µM); SphK2 (IC50=3.2 µM)]. Our results could contribute to the development of novel potent and/or selective inhibitors via modifications of lipid substrates.
1H-NMR spectra were recorded using a JEOL ECA-500 spectrometer at 500 MHz frequency. Chemical shifts are reported in δ (ppm) relative to Me4Si (in CDCl3) as internal standard. 13C-NMR spectra were recorded using a JEOL ECA-500 and referenced to the residual CHCl3 signal. Exact mass (HR-MS) spectra were recorded on a Shimadzu LC-ESI-IT-TOF-MS equipment (electrospray ionization (ESI)). Optical rotations were measured with a JASCO P-1020 polarimeter. For column chromatography, Wakogel C-300E (Wako, Osaka, Japan) or Chromatorex NH-DM1020 (Fuji Silysia, Japan) was employed.
Benzyl [(3R,4S,5S)-4-Hydroxy-5-vinyltetrahydrofuran-3-yl]carbamate (4)To a mixture of 3 (300 mg, 1.31 mmol) in dioxane (1.0 mL) was added 4 M HCl/dioxane (4.0 mL). The mixture was stirred at room temperature until disappearance of the starting material (monitored by TLC). The mixture was evaporated in vacuo to give a crude product, which was used without further purification. To a mixture of the crude product in CH2Cl2 (3.0 mL) were added Et3N (720 µL, 5.24 mmol) and Cbz-Cl (367 µL, 2.62 mmol) at 0°C. The mixture was stirred at room temperature for 3 h. After completion of the reaction, the reaction mixture was diluted with EtOAc and washed with saturated aqueous NaHCO3 and brine. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to give a crude product, which was purified by column chromatography on silica gel with hexane/EtOAc (1 : 1) to give 4 (124 mg, 36%) as a white solid: mp 92–93°C; [α]D29+19.0 (c=0.83, CHCl3); 1H-NMR (CDCl3) δ: 2.86–2.88 (1H, m), 3.63 (1H, dd, J=9.2, 2.3 Hz), 4.11–4.16 (1H, m), 4.16–4.18 (1H, m), 4.28 (1H, dd, J=9.2, 5.7 Hz), 4.44 (1H, dd, J=5.7, 4.6 Hz), 5.07 (1H, d, J=12.0 Hz), 5.11 (1H, d, J=12.0 Hz), 5.19 (1H, d, J=5.2 Hz), 5.33 (1H, d, J=10.9 Hz), 5.42 (1H, d, J=17.2 Hz), 5.89–5.95 (1H, m), 7.30–7.37 (5H, m); 13C-NMR (CDCl3) δ: 59.6, 67.1, 70.7, 77.0, 81.8, 118.5, 128.2, 128.3 (2C), 128.6 (2C), 132.7, 136.0, 156.0; ESI-TOF-MS m/z: 286.1046 (Calcd for C14H17NNaO4: 286.1050).
1-(But-3-en-1-yloxy)nonane (5a)To a mixture of 3-buten-1-ol (200 mg, 2.77 mmol) in N,N-dimethylformamide (DMF) (2.0 mL) was added 1-bromononane (355 µL, 1.85 mmol) and sodium hydride (NaH) (60% in mineral oil, 112 mg, 2.77 mmol) at room temperature. The mixture was stirred at room temperature until disappearance of the starting material (monitored by TLC). The reaction was quenched by H2O. The whole was extracted with EtOAc/hexane (10 : 1). The organic layer was washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo to give a crude product, which was purified by column chromatography on silica gel with hexane/EtOAc (20 : 1) to give 5a (249 mg, 66%) as a colorless oil. The 1H- and 13C-NMR spectra were in agreement with those reported by our group25): 1H-NMR (CDCl3) δ: 0.88 (3H, t, J=6.9 Hz), 1.18–1.41 (12H, m), 1.54–1.59 (2H, m), 2.31–2.36 (2H, m), 3.41 (2H, t, J=6.9 Hz), 3.46 (2H, t, J=6.9 Hz), 5.03 (1H, dd, J=10.3, 1.1 Hz), 5.09 (1H, dt, J=17.2, 1.7 Hz), 5.79–5.87 (1H, m); 13C-NMR (CDCl3) δ: 14.1, 22.7, 26.1, 29.3, 29.48, 29.54, 29,7, 31.9, 34.2, 70.1, 71.0, 116.2, 135.3.
1-(Hex-5-en-1-yloxy)heptane (5b)According to the procedure described for the preparation of 5a, 1-bromoheptane (299 µL, 1.85 mmol) was converted into 5b (136 mg, 36%) by the reaction with 5-hexen-1-ol (330 µL, 2.77 mmol) and NaH (60% in mineral oil, 112 mg, 2.77 mmol). The 1H- and 13C-NMR spectra were in agreement with those reported by our group25): colorless oil; 1H-NMR (CDCl3) δ: 0.88 (3H, t, J=6.9 Hz), 1.26–1.33 (8H, m), 1.42–1.48 (2H, m), 1.55–1.60 (4H, m), 2.06–2.10 (2H, m), 3.39 (2H, t, J=6.3 Hz), 3.40 (2H, t, J=6.3 Hz), 4.94 (1H, dd, J=10.3, 1.1 Hz), 5.00 (1H, dd, J=17.2, 1.7 Hz), 5.77–5.85 (1H, m); 13C-NMR (CDCl3) δ: 14.1, 22.6, 25.5, 26.1, 29.16, 29.21, 29,8, 31.8, 33.6, 70.7, 71.0, 114.4, 138.8.
8-(Pentyloxy)oct-1-ene (5c)According to the procedure described for the preparation of 5a, 1-bromopentane (237 µL, 1.85 mmol) was converted into 5c (225 mg, 60%) by the reaction with 7-octen-1-ol (422 µL, 2.77 mmol) and NaH (60% in mineral oil, 112 mg, 2.77 mmol): colorless oil; 1H-NMR (CDCl3) δ: 0.90 (3H, t, J=6.9 Hz), 1.28–1.42 (10H, m), 1.52–1.60 (4H, m), 2.02–2.06 (2H, m), 3.39 (4H, t, J=6.9 Hz), 4.90–4.95 (1H, m), 4.97–5.01 (1H, m), 5.76–5.86 (1H, m); 13C-NMR (CDCl3) δ: 14.0, 22.5, 26.0, 28.3, 28.8, 28.9, 29.5, 29.7, 33.7, 70.88, 70.93, 114.1, 139.1; Anal. Calcd for C13H26O: C, 78.72; H, 13.21. Found: C, 78.58, H, 13.16.
10-Propoxydec-1-ene (5d)According to the procedure described for the preparation of 5a, 1-bromopropane (172 µL, 1.85 mmol) was converted into 5d (209 mg, 55%) by the reaction with 9-decen-1-ol (515 µL, 2.77 mmol) and NaH (60% in mineral oil, 112 mg, 2.77 mmol): colorless oil; 1H-NMR (CDCl3) δ: 0.91 (3H, t, J=6.9 Hz), 1.24–1.40 (10H, m), 1.56–1.61 (4H, m), 2.01–2.07 (2H, m), 3.36 (2H, t, J=6.9 Hz), 3.39 (2H, t, J=6.6 Hz), 4.91–4.94 (1H, m), 4.97–5.01 (1H, m), 5.77–5.85 (1H, m); 13C-NMR (CDCl3) δ: 10.6, 22.9, 26.2, 28.9, 29.1, 29.4 (2C), 29,8, 33.8, 70.9, 72.5, 114.1, 139.2; Anal. Calcd for C13H26O: C, 78.72; H, 13.21. Found: C, 78.59, H, 13.13.
12-Methoxydodec-1-ene (5e)According to the procedure described for the preparation of 5a, 12-bromododec-1-ene (470 mg, 1.85 mmol) was converted into 5e (286 mg, 76%) by the reaction with methanol (114 µL, 2.77 mmol) and NaH (60% in mineral oil, 112 mg, 2.77 mmol): colorless oil; 1H-NMR (CDCl3) δ: 1.27–1.36 (12H, m), 1.32–1.39 (2H, m), 1.53–1.59 (2H, m), 2.01–2.06 (2H, m), 3.32 (3H, s), 3.35 (2H, t, J=6.6 Hz), 4.89–4.93 (1H, m), 4.96–5.00 (1H, m), 5.76–5.84 (1H, m); 13C-NMR (CDCl3) δ: 26.1, 28.9, 29.1, 29.4 (2C), 29.47, 29.52, 29.6, 33.7, 58.4, 72.9, 114.0, 139.0; Anal. Calcd for C13H26O: C, 78.72; H, 13.21. Found: C, 78.66, H, 13.38.
(2S,3S,4R)-4-Amino-2-[4-(nonyloxy)butyl]tetrahydrofuran-3-ol (1a)To a stirred mixture of 4 (20.0 mg, 0.0760 mmol) and 5a (45.2 mg, 0.228 mmol) in CH2Cl2 (3.0 mL) was added 2nd gen. Hoveyda–Grubbs catalyst (4.8 mg, 0.0076 mmol) at room temperature. The mixture was stirred at 45°C until disappearance of the starting material (monitored by TLC). The reaction mixture was filtered through a short pad of Chromatorex NH-DM1020, and then the filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel (hexane/EtOAc=3 : 2) to afford an E/Z mixture of alkene (21.4 mg), which was used without further purification. To a mixture of the alkene (10.0 mg) in CH2Cl2/EtOH (1 : 1, 3.0 mL) was added Pd(OH)2/C (ca. 50% wt on carbon, 3.2 mg, 0.016 mmol). The mixture was stirred overnight at room temperature under H2 atmosphere (0.5 MPa). The reaction mixture was filtered through a 0.5 µm polytetrafluoroethylene (PTFE) membrane filter (Advantec), and then the filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel (CHCl3/MeOH/NH4OH=90 : 9 : 1) to afford 1a (7.1 mg, 53%) as a white waxy solid: [α]D28+1.2 (c=0.20, MeOH); 1H-NMR (CD3OD) δ: 0.89 (3H, t, J=6.9 Hz), 1.28–1.34 (12H, m), 1.39–1.65 (8H, m), 3.29–3.32 (1H, m), 3.38 (1H, dd, J=9.2, 3.4 Hz), 3.41 (2H, t, J=6.3 Hz), 3.43 (2H, t, J=6.3 Hz), 3.80–3.82 (1H, m), 3.89 (1H, ddd, J=6.9, 6.9, 3.4 Hz), 4.12 (1H, dd, J=9.2, 5.7 Hz); 13C-NMR (CD3OD) δ: 14.4, 23.7, 24.1, 27.3, 29.5, 30.4, 30.6, 30.7, 30.8, 30.9, 33.1, 60.8, 71.8, 72.0, 74.0, 79.6, 82.3; ESI-TOF-MS m/z: 302.2694 (Calcd for C17H36NO3: 302.2690).
(2S,3S,4R)-4-Amino-2-[6-(heptyloxy)hexyl]tetrahydrofuran-3-ol (1b)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (20.0 mg, 0.0760 mmol) was converted into an E/Z mixture of alkene (21.4 mg) by the reaction with 5b (45.2 mg, 0.228 mmol) and 2nd gen. Hoveyda–Grubbs catalyst (4.8 mg, 0.0076 mmol). The alkene (10.0 mg) was converted into 1b (6.1 mg, 57%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 3.2 mg, 0.016 mmol): white waxy solid; [α]D28+2.8 (c=0.18, MeOH); 1H-NMR (CD3OD) δ: 0.90 (3H, t, J=7.2 Hz), 1.30–1.43 (14H, m), 1.52–1.60 (6H, m), 3.31–3.33 (1H, m), 3.37–3.39 (1H, m), 3.41 (2H, t, J=6.9 Hz), 3.41 (2H, t, J=6.9 Hz), 3.80–3.81 (1H, m), 3.88 (1H, ddd, J=6.9, 6.9, 3.4 Hz), 4.12 (1H, dd, J=9.2, 5.7 Hz); 13C-NMR (CD3OD) δ: 14.4, 23.7, 27.3, 27.4, 29.7, 30.3, 30.7, 30.8 (3C), 33.0, 60.8, 71.92, 71.94, 73.9, 79.6, 82.3; ESI-TOF-MS m/z: 302.2683 (Calcd for C17H36NO3: 302.2690).
(2S,3S,4R)-4-Amino-2-[8-(pentyloxy)octyl]tetrahydrofuran-3-ol (1c)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (20.0 mg, 0.0760 mmol) was converted into an E/Z mixture of alkene (25.2 mg) by the reaction with 5c (45.2 mg, 0.228 mmol) and 2nd gen. Hoveyda–Grubbs catalyst (4.8 mg, 0.0076 mmol). The alkene (10.0 mg) was converted into 1c (7.1 mg, 78%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 3.2 mg, 0.016 mmol): white waxy solid; [α]D28+0.71 (c=0.25, MeOH); 1H-NMR (CD3OD) δ: 0.91 (3H, t, J=6.9 Hz), 1.32–1.42 (14H, m), 1.52–1.62 (6H, m), 3.31–3.33 (1H, m), 3.38 (1H, dd, J=9.2, 3.4 Hz), 3.41 (2H, t, J=6.3 Hz), 3.41 (2H, t, J=6.3 Hz), 3.80–3.81 (1H, m), 3.88 (1H, ddd, J=6.9, 6.9, 3.4 Hz), 4.12 (1H, dd, J=9.2, 5.7 Hz); 13C-NMR (CD3OD) δ: 14.4, 23.6, 27.3, 27.5, 29.5, 29.7, 30.46, 30.52, 30.7, 30.8, 30.9, 60.8, 71.9 (2C), 73.9, 79.6, 82.4; ESI-TOF-MS m/z: 302.2694 (Calcd for C17H36NO3: 302.2690).
(2S,3S,4R)-4-Amino-2-(10-propoxydecyl)tetrahydrofuran-3-ol (1d)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (20.0 mg, 0.0760 mmol) was converted into an E/Z mixture of alkene (24.6 mg) by the reaction with 5d (45.2 mg, 0.228 mmol) and 2nd gen. Hoveyda-Grubbs catalyst (4.8 mg, 0.0076 mmol). The alkene (12.0 mg) was converted into 1d (7.3 mg, 65%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 3.8 mg, 0.019 mmol): white waxy solid; [α]D28+1.9 (c=0.28, MeOH); 1H-NMR (CD3OD) δ: 0.92 (3H, t, J=6.9 Hz), 1.31–1.45 (14H, m), 1.53–1.61 (6H, m), 3.30–3.33 (1H, m), 3.37–3.39 (1H, m), 3.37 (2H, t, J=6.6 Hz), 3.41 (2H, t, J=6.6 Hz), 3.80–3.80 (1H, m), 3.88 (1H, ddd, J=6.6, 6.6, 3.4 Hz), 4.12 (1H, dd, J=9.7, 5.7 Hz); 13C-NMR (CD3OD) δ: 10.9, 23.9, 27.3, 27.5, 29.7, 30.6, 30.67, 30.70, 30.73, 30.8, 30.9, 60.9, 71.9, 73.6, 74.0, 79.6, 82.4; ESI-TOF-MS m/z: 302.2699 (Calcd for C17H36NO3: 302.2690).
(2S,3S,4R)-4-Amino-2-(12-methoxydodecyl)tetrahydrofuran-3-ol (1e)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (20.0 mg, 0.0760 mmol) was converted into an E/Z mixture of alkene (19.6 mg) by the reaction with 5e (45.2 mg, 0.228 mmol) and 2nd gen. Hoveyda–Grubbs catalyst (4.8 mg, 0.0076 mmol). The alkene (10.0 mg) was converted into 1e (6.1 mg, 65%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 3.2 mg, 0.016 mmol): white waxy solid; [α]D29+2.1 (c=0.11, MeOH); 1H-NMR (CD3OD) δ: 1.29–1.42 (18H, m), 1.53–1.58 (4H, m), 3.30 (3H, s), 3.30–3.32 (1H, m), 3.36–3.39 (1H, m), 3.37 (2H, t, J=6.9 Hz), 3.79–3.80 (1H, m), 3.87 (1H, ddd, J=6.7, 6.7, 3.6 Hz), 4.12 (1H, dd, J=9.2, 5.7 Hz); 13C-NMR (CD3OD) δ: 23.9, 27.2, 27.5, 29.7, 30.58, 30.60, 30.7 (3C), 30.8, 30.9, 58.7, 60.9, 73.9, 74.0, 79.6, 82.4; ESI-TOF-MS m/z: 302.2688 (Calcd for C17H36NO3: 302.2690).
Benzyl But-3-en-1-yl(nonyl)carbamate (6a)To a mixture of but-3-en-1-amine (500 mg, 7.03 mmol) in CH2Cl2 (15 mL) were added Et3N (970 µL, 7.03 mmol) and Cbz-Cl (793 µL, 5.62 mmol) at 0°C. The mixture was stirred at room temperature until disappearance of the starting material (monitored by TLC). After being stirred overnight, the reaction mixture was diluted with CH2Cl2 and washed with 1 M HCl, saturated aqueous NaHCO3 and brine. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to give a crude product (1.18 g), which was used without further purification. To a mixture of the crude product (200 mg) in DMF (1.0 mL) were added 1-bromononane (277 µL, 1.46 mmol) and NaH (60% in mineral oil, 58.4 mg, 1.46 mmol) at room temperature. The mixture was stirred at room temperature overnight. After completion of the reaction, the reaction was quenched by H2O. The whole was extracted with EtOAc/hexane (10 : 1). The organic layer was washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo to give a crude product, which was purified by column chromatography on silica gel with hexane/EtOAc (20 : 1) to give 6a (231 mg, 56%) as a colorless oil: 1H-NMR (CDCl3) δ: 0.88 (3H, t, J=7.4 Hz), 1.23–1.29 (12H, m), 1.49–1.55 (2H, m), 2.26–2.32 (2H, m), 3.20–3.33 (4H, m), 4.99–5.08 (2H, m), 5.13 (2H, s), 5.71–5.79 (1H, m), 7.28–7.37 (5H, m); 13C-NMR (CDCl3, ca. 1 : 1 mixture of rotamers) δ: 14.1, 22.6, 26.8, 28.1 (0.5C), 28.6 (0.5C), 29.2, 29.3 (0.5C), 29.4 (0.5C), 29.5, 31.8, 32.6 (0.5C), 33.2 (0.5C), 46.4 (0.5C), 47.0 (0.5C), 47.1 (0.5C), 47.8 (0.5C), 66.8, 116.5 (0.5C), 116.6 (0.5C), 127.7 (2C), 127.8, 128.4 (2C), 135.2 (0.5C), 135.4 (0.5C), 137.0, 156.0; ESI-TOF-MS m/z: 332.2588 (Calcd for C21H34NO2: 332.2584).
Benzyl Heptyl(hex-5-en-1-yl)carbamate (6b)According to the procedure described for the preparation of 6a, heptylamine (1.00 g, 8.68 mmol) was converted into the crude Cbz-protected amine (1.35 g) by the reaction with Et3N (1.20 mL, 8.68 mmol) and Cbz-Cl (979 µL, 6.94 mmol). The crude Cbz-protected amine (200 mg) was converted into 6b (171 mg, 40%) by the reaction with 6-bromo-1-hexene (160 µL, 1.20 mmol) and NaH (60% in mineral oil, 48.0 mg, 1.20 mmol): colorless oil; 1H-NMR (CDCl3) δ: 0.87 (3H, t, J=6.9 Hz), 1.24–1.40 (10H, m), 1.50–1.56 (4H, m), 2.01–2.10 (2H, m), 3.19–3.26 (4H, m), 4.93–5.02 (2H, m), 5.12 (2H, s), 5.74–5.81 (1H, m), 7.27–7.38 (5H, m); 13C-NMR (CDCl3, ca. 1 : 1 mixture of rotamers) δ: 14.0, 22.5, 26.0, 26.7, 27.5 (0.5C), 28.0 (0.5C), 28.1 (0.5C), 28.6 (0.5C), 29.0 (0.5C), 29.1 (0.5C), 31.7, 33.4 (0.5C), 33.5 (0.5C), 46.7 (0.5C), 46.9 (0.5C), 47.3 (0.5C), 47.5 (0.5C), 66.7, 114.6, 127.7 (2C), 127.8, 128.5 (2C), 137.1, 138.4 (0.5C), 138.6 (0.5C), 156.1; ESI-TOF-MS m/z: 332.2589 (Calcd for C21H34NO2: 332.2584).
Benzyl Oct-7-en-1-yl(pentyl)carbamate (6c)According to the procedure described for the preparation of 6a, amylamine (1.00 g, 11.4 mmol) was converted into the crude Cbz-protected amine (1.60 g) by the reaction with Et3N (1.58 mL, 11.4 mmol) and Cbz-Cl (1.29 mL, 9.12 mmol). The crude Cbz-protected amine (200 mg) was converted into 6c (171 mg, 40%) by the reaction with 8-bromo-1-octene (226 µL, 1.35 mmol) and NaH (60% in mineral oil, 54.0 mg, 1.35 mmol): colorless oil; 1H-NMR (CDCl3) δ: 0.86–0.89 (3H, m), 1.24–1.34 (10H, m), 1.50–1.55 (4H, m), 2.00–2.05 (2H, m), 3.19–3.25 (4H, m), 4.91–4.94 (1H, m), 4.98 (1H, d, J=17.2 Hz), 5.12 (2H, s), 5.76–5.82 (1H, m), 7.26–7.38 (5H, m); 13C-NMR (CDCl3, ca. 1 : 1 mixture of rotamers) δ: 14.0, 22.4, 26.6, 27.8 (0.5C), 28.0 (0.5C), 28.3 (0.5C), 28.5 (0.5C), 28.7, 28.8, 28.9, 33.7, 46.8, 47.5, 66.7, 114.2, 127.7 (2C), 127.7, 128.4 (2C), 137.1, 138.9, 156.1; ESI-TOF-MS m/z: 332.2581 (Calcd for C21H34NO2: 332.2584).
Benzyl Dec-9-en-1-yl(propyl)carbamate (6d)According to the procedure described for the preparation of 6a, propylamine (1.00 g, 16.9 mmol) was converted into the crude Cbz-protected amine (1.90 g) by the reaction with Et3N (2.30 mL, 16.9 mmol) and Cbz-Cl (1.90 mL, 13.5 mmol). The crude Cbz-protected amine (200 mg) was converted into 6d (179 mg, 30%) by the reaction with 10-bromo-1-decene (310 µL, 1.55 mmol) and NaH (60% in mineral oil, 62.0 mg, 1.55 mmol): colorless oil; 1H-NMR (CDCl3) δ: 0.85–0.89 (3H, m), 1.24–1.35 (10H, m), 1.50–1.57 (4H, m), 2.01–2.06 (2H, m), 3.18–3.24 (4H, m), 4.93 (1H, d, J=10.3 Hz), 4.99 (1H, dt, J=18.9, 1.7 Hz), 5.12 (2H, s), 5.76–5.85 (1H, m), 7.28–7.36 (5H, m); 13C-NMR (CDCl3, ca. 1 : 1 mixture of rotamers) δ: 11.2, 21.3 (0.5C), 21.8 (0.5C), 26.8, 28.1 (0.5C), 28.6 (0.5C), 28.8, 29.0, 29.25, 29.34, 33.7, 46.9 (0.5C), 47.6 (0.5C), 48.5 (0.5C), 49.1 (0.5C), 66.7, 114.1, 127.6 (2C), 128.3, 128.3 (2C), 137.1, 139.1, 156.1; ESI-TOF-MS m/z: 332.2586 (Calcd for C21H34NO2: 332.2584).
Benzyl Dodec-11-en-1-yl(methyl)carbamate (6e)According to the procedure described for the preparation of 6a, methylamine (2.0 M in tetrahydrofuran, 5.00 mL, 10.0 mmol) was converted into the crude Cbz-protected amine (1.18 g) by the reaction with Et3N (1.39 mL, 10.0 mmol) and Cbz-Cl (1.13 mL, 8.00 mmol). The crude Cbz-protected amine (200 mg) was converted into 6e (282 mg, 50%) by the reaction with 12-bromo-1-dodecene (449 mg, 1.80 mmol) and NaH (60% in mineral oil, 72.0 mg, 1.80 mmol): colorless oil; 1H-NMR (CDCl3) δ: 1.23–1.29 (12H, m), 1.34–1.40 (2H, m), 1.48–1.54 (2H, m), 2.01–2.06 (2H, m), 2.90 (3H, s), 3.26 (2H, q, J=9.1 Hz), 4.92–4.94 (1H, m), 4.97–5.01 (1H, m), 5.12 (2H, s), 5.77–5.85 (1H, m), 7.27–7.37 (5H, m); 13C-NMR (CDCl3, ca. 1 : 1 mixture of rotamers) δ: 14.1, 26.6, 27.4 (0.5C), 27.8 (0.5C), 28.6, 29.1, 29.39, 29.44, 33.7, 33.8 (0.5C), 34.5 (0.5C), 48.6, 49.1, 66.8, 114.0, 127.7 (2C), 127.8, 128.3 (2C), 137.1, 139.1, 156.2; ESI-TOF-MS m/z: 332.2580 (Calcd for C21H34NO2: 332.2584).
(2S,3S,4R)-4-Amino-2-[4-(nonylamino)butyl]tetrahydrofuran-3-ol (2a)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (22.0 mg, 0.0836 mmol) was converted into an E/Z mixture of alkene (36.0 mg) by the reaction with 6a (83.1 mg, 0.251 mmol) and 2nd gen. Hoveyda–Grubbs catalyst (5.3 mg, 0.0084 mmol). The alkene (17.0 mg) was converted into 2a (10.2 mg, 86%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 4.2 mg, 0.015 mmol): white waxy solid; [α]D27−10.2 (c=0.37, MeOH); 1H-NMR (CD3OD) δ: 0.89 (3H, t, J=6.9 Hz), 1.29–1.37 (12H, m), 1.45–1.61 (2H, m), 1.68–1.75 (6H, m), 2.96–3.01 (4H, m), 3.61–3.64 (2H, m), 3.95 (1H, ddd, J=6.4, 6.4, 4.2 Hz), 4.19–4.24 (2H, m); 13C-NMR (CD3OD) δ: 14.4, 23.7, 24.4, 27.30, 27.33, 27.6, 28.9, 30.2, 30.3, 30.5, 33.0, 49.0, 49.2, 59.8, 69.4, 75.6, 82.4; ESI-TOF-MS m/z: 301.2850 (Calcd for C17H37N2O2: 301.2850).
(2S,3S,4R)-4-Amino-2-[6-(heptylamino)hexyl]tetrahydrofuran-3-ol (2b)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (25.0 mg, 0.0960 mmol) was converted into an E/Z mixture of alkene (33.1 mg) by the reaction with 6b (94.4 mg, 0.285 mmol) and 2nd gen. Hoveyda–Grubbs catalyst (6.0 mg, 0.0095 mmol). The alkene (16.0 mg) was converted into 2b (11.2 mg, 83%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 4.0 mg, 0.014 mmol): white waxy solid; [α]D27−9.9 (c=0.43, MeOH); 1H-NMR (CD3OD) δ: 0.82 (3H, t, J=6.9 Hz), 1.19–1.44 (14H, m), 1.52–1.62 (6H, m), 2.85–2.91 (4H, m), 3.49–3.56 (2H, m), 3.83 (1H, ddd, J=6.8, 6.8, 4.0 Hz), 4.09–4.15 (2H, m); 13C-NMR (CD3OD) δ: 14.4, 23.6, 27.0, 27.2, 27.3, 27.5, 27.6, 29.2, 29.9, 30.2, 32.7, 49.0 (2C), 59.8, 69.4, 75.6, 82.7; ESI-TOF-MS m/z: 301.2850 (Calcd for C17H37N2O2: 301.2850).
(2S,3S,4R)-4-Amino-2-[8-(pentylamino)octyl]tetrahydrofuran-3-ol (2c)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (25.0 mg, 0.0950 mmol) was converted into an E/Z mixture of alkene (38.2 mg) by the reaction with 6c (94.4 mg, 0.285 mmol) and 2nd gen. Hoveyda–Grubbs catalyst (6.0 mg, 0.0095 mmol). The alkene (19.0 mg) was converted into 2c (15.8 mg, 99%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 4.5 mg, 0.016 mmol): white waxy solid; [α]D27−9.5 (c=0.45, MeOH); 1H-NMR (CD3OD) δ: 0.85 (3H, t, J=6.9 Hz), 1.24–1.40 (14H, m), 1.50–1.54 (2H, m), 1.57–1.62 (4H, m), 2.86–2.90 (4H, m), 3.50–3.54 (2H, m), 3.81 (1H, ddd, J=6.7, 6.7, 4.2 Hz), 4.09–4.10 (1H, m), 4.12 (1H, dd, J=11.4, 7.4 Hz); 13C-NMR (CD3OD) δ: 14.1, 23.2, 27.0, 27.26, 27.30, 27.6, 29.4, 29.7, 30.1, 30.4, 30.7, 49.0 (2C), 59.8, 69.4, 75.6, 82.8; ESI-TOF-MS m/z: 301.2840 (Calcd for C17H37N2O2: 301.2850).
(2S,3S,4R)-4-Amino-2-[10-(propylamino)decyl]tetrahydrofuran-3-ol (2d)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (24.4 mg, 0.0927 mmol) was converted into an E/Z mixture of alkene (37.4 mg) by the reaction with 6d (88.6 mg, 0.278 mmol) and 2nd gen. Hoveyda-Grubbs catalyst (5.8 mg, 0.0093 mmol). The alkene (16.0 mg) was converted into 2d (10.7 mg, 73%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 4.1 mg, 0.015 mmol): white waxy solid; [α]D26−9.4 (c=0.43, MeOH); 1H-NMR (CD3OD) δ: 1.01 (3H, t, J=7.4 Hz), 1.31–1.39 (14H, m), 1.59–1.62 (2H, m), 1.67–1.73 (4H, m), 2.93–2.98 (4H, m), 3.59–3.62 (2H, m), 3.89 (1H, ddd, J=6.7, 6.7, 4.2 Hz), 4.18–4.19 (1H, m), 4.21 (1H, dd, J=11.2, 7.2 Hz); 13C-NMR (CD3OD) δ: 11.2, 20.7, 27.29, 27.31, 27.6, 29.4, 30.1, 30.5, 30.57, 30.64, 30.8, 49.0, 50.5, 59.8, 69.3, 75.6, 82.8; ESI-TOF-MS m/z: 301.2859 (Calcd for C17H37N2O2: 301.2850).
(2S,3S,4R)-4-Amino-2-[12-(methylamino)dodecyl]tetrahydrofuran-3-ol (2e)According to the procedure described for the preparation of 1a, the Cbz-protected amine 4 (25.0 mg, 0.0949 mmol) was converted into an E/Z mixture of alkene (37.6 mg) by the reaction with 6e (94.4 mg, 0.285 mmol) and 2nd gen. Hoveyda-Grubbs catalyst (6.0 mg, 0.0095 mmol). The alkene (17.0 mg) was converted into 2e (11.2 mg, 89%) by the reaction with Pd(OH)2/C (ca. 50% wt on carbon, 4.1 mg, 0.015 mmol): white waxy solid; [α]D27−8.4 (c=0.83, MeOH); 1H-NMR (CD3OD) δ: 1.21–1.31 (18H, m), 1.51–1.59 (4H, m), 2.59 (3H, s), 2.86–2.90 (2H, m), 3.50–3.54 (2H, m), 3.81 (1H, ddd, J=6.3, 6.3, 4.0 Hz), 4.09–4.10 (1H, m), 4.13 (1H, dd, J=11.4, 7.4 Hz); 13C-NMR (CD3OD) δ: 27.2, 27.3, 27.5, 29.4, 30.2, 30.5, 30.6, 30.70, 30.72, 30.73, 30.9, 33.6, 50.5, 59.8, 69.3, 75.6, 82.8; ESI-TOF-MS m/z: 301.2855 (Calcd for C17H37N2O2: 301.2850).
SphK inhibitory activities were evaluated by the off-chip mobility shift assay by the QuickScout service from Carna Bioscience (Kobe, Japan). SphK1 (1–384) and SphK2 (1–618) were expressed as N-terminal glutathione S transferase (GST)-fusion proteins using a baculovirus expression system. They were purified using glutathione Sepharose chromatography. Each chemical in dimethyl sulfoxide (DMSO) at different concentrations was diluted fourfold with reaction buffer [20 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES) (pH 7.5), 0.01% Triton X-100, 2 mM dithiothreitol (DTT)]. For SphK reactions, a combination of the compound, 1 µM sphingosine, 5 mM MgCl2, ATP (25 µM for SphK1; 600 µM for SphK2) in reaction buffer (20 µL) were incubated with each SphK in 384-well plates at room temperature for 1 h (n=2). The reaction was terminated by addition of 60 or 70 µL of termination buffer (Carna Biosciences). Substrate and product were separated by electrophoretic means using the LabChip system. The kinase reaction was evaluated by the product ratio, which was calculated from the peak heights of the substrate (S) and product (P): [P/(P+S)]. Inhibition data were calculated by comparing with noenzyme controls for 100% inhibition and no-inhibitor reactions for 0% inhibition. IC50 values were calculated using GraphPad Prism 7 software (GraphPad Software, Incorporated, La Jolla, CA, U.S.A.).
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Numbers JP15KT0061, JP15H04654, JP16K16638 and JP18H04615), and Japan Agency for Medical Research and Development (AMED) under Grant Number JP18gm1010007 and JP18ak0101072.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials (details of computations).