Regular Articles
Spontaneous and Direct Transformation of N,O-Diaryl Carbamates into N,N′-Diarylureas
2018 年 66 巻 9 号 p. 880-884
詳細
2018 年 66 巻 9 号 p. 880-884
We have discovered a spontaneous reaction of N,O-diaryl carbamates to afford symmetrical N,N′-diarylureas. Optimization of the conditions indicated that N,N-dimethylformamide (DMF) was the best solvent and triethylamine (Et3N) was the best additive for this transformation. The reaction requires the presence of aryl groups on the nitrogen and oxygen atoms of carbamates. Substrates bearing an electron-donating methoxy group on either of the aryl groups reacted slowly under these conditions.
The N,N′-diphenylurea skeleton is found in many biologically active compounds, including cytokinin,1) antischistosomal agent,2) diacylglycerol acyltransferase (DGAT)1 inhibitor,3) vascular endothelial growth factor receptor (VEGFR)-2 inhibitor,4) BKca channel activator,5) and p-38-mitogen-activated protein (MAP) kinase inhibitor.6) In addition, N,N′-diphenyl-substituted urea derivatives are useful components for gel-forming materials7) and crystal engineering.8)
N,N′-Diphenylurea derivatives are typically synthesized by reaction of amine with aryl isocyanates or phosgene,9) or with carbamates.10–14) Indeed, unsymmetrically substituted ureas can be prepared from carbamate by direct reaction with amine.13–15) Analogously with the reaction of carbamates, N-arylcarbamates decompose to afford phenyl isocyanate in the presence of triethylamine (Et3N) via the E1cB mechanism10) (Fig. 1a). In addition, phenyl isocyanate affords N,N′-diphenylurea even in the absence of amine16–18) (Fig. 1b). However, carbamates have been known as a “blocked isocyanate,”19) there are few examples of the synthesis of urea compounds via carbamate with a single-pot operation (Fig. 1c); a precedent required high temperature (200°C) and did not report isolated yield of urea.20) Such reactions would be useful, considering the difficulty of handling highly reactive aryl isocyanates. Here, we report a spontaneous, single-step formation of symmetrical N,N′-diarylurea compounds from N,O-diaryl carbamates.

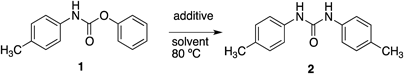
To examine the direct transformation of N,O-diaryl carbamate to symmetrical N,N′-diarylurea, we chose N-(4-methylphenyl)-O-phenyl carbamate (1) as a model substrate (Table 1). We were pleased to find that 1 afforded N,N′-bis(4-methylphenyl)urea (2) in 91% yield in the presence of 1 equiv of Et3N in N,N-dimethylformamide (DMF) (Table 1, entry 1) in open-air environment.21) When the reaction was performed at 50°C, the yield of 2 was decreased (Table 1 entry 2). Other less polar solvents, such as 1,4-dioxane, toluene, and dichloroethane also resulted in lower yield (Table 1, entries 3–5).
 | ||||
|---|---|---|---|---|
| Entry | Additive (eq) | Solvent | Time (h) | Yield of 2 (%)a) |
| 1 | Et3N (1) | DMF | 25 | 91 |
| 2b) | Et3N (1) | DMF | 23 | 71 |
| 3 | Et3N (1) | 1,4-Dioxane | 96 | 55 |
| 4 | Et3N (1) | Toluene | 121 | 28 |
| 5 | Et3N (1) | ClCH2CH2Cl | 65 | 45 |
| 6 | Et3N (1) | DMSO | 32 | 32 |
| 7 | Et3N (1) | DMA | 52 | 38 |
| 8 | Et3N (1) | NMP | 44 | 45 |
| 9 | Et3N (1) | DMI | 30 | 95 |
| 10 | Et3N (0.01) | DMF | 24 | 93 |
| 11 | None | DMF | 70 | 82 |
| 12 | DMAP (0.01) | DMF | 116 | 75 |
| 13 | 2,6-Lutidine (0.01) | DMF | 71 | 89 |
| 14 | Imidazole (0.01) | DMF | 67 | 83 |
| 15 | TMEDA (0.01) | DMF | 50 | 95 |
a) Isolated yield. Yields were calculated based on the assumption that 2 eq of aryl carbamate was consumed to generate 1 eq of aryl urea. b) The reaction was performed at 50°C.
The use of dimethyl sulfoxide (DMSO) as the solvent resulted in the formation of a by-product, presumed to be the product of trimerization of isocyanate, and the yield of the desired 2 was only 32% (Table 1, entry 6). Among other polar non-protic solvents, 1,3-dimethylimidazolidinone (DMI) afforded 2 in 95% yield (Table 1, entries 7–9) after a slightly longer reaction time. The reaction proceeded even with only a catalytic amount of Et3N (1 mol %), and afforded 2 in comparable yield (Table 1, entry 10). The role of Et3N in accelerating the reaction was highlighted by the reaction in the absence of Et3N, which required a longer reaction time and afforded only moderate yield (Table 1, entry 11). Other amine additives, such as N,N-dimethylaminopyridine (DMAP), 2,6-lutidine, imidazole, and tetramethylethylenediamine (TMEDA), were less effective than Et3N, and required a longer reaction time to attain a reasonable product yield (Table 1, entries 12–15).
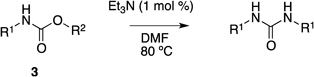
With the optimized conditions in hand, we explored the scope of the reaction (Table 2). When the reaction was performed with 3a, which has an electron-withdrawing nitro group on the anilino group, the outcome of the reaction was comparable with that of 1 (entry 1). In contrast, the carbamate bearing an electron-donating methoxy group on the anilino group, 3b, reacted slowly under the same conditions, and the yield was moderate (entry 2). Replacement of the anilino moiety with a benzyl group impeded the reaction, and the desired diphenylurea product was not obtained (3c, entry 3); most of the starting material was recovered. This result shows the importance of the phenyl moiety for this direct transformation. Replacement of the O-aryl moiety with various substituted phenyl groups affected the yield of the corresponding symmetrical diphenyl urea similarly; introduction of an electron-withdrawing nitro group (3d) afforded a good yield of N,N′-diphenylurea in a shorter reaction time, while the electron-donating methoxy group (3e) required a longer reaction time to afford a comparable yield of the urea compound. When the O-aryl group was replaced with a methyl group (3f), only a small amount of N,N′-diphenylurea was obtained and most of the starting material, carbamate 3f, remained intact. These results indicate that the presence of an aryl group on both nitrogen and oxygen of carbamate is required, and there is a preference for an electron-withdrawing substituent on the phenyl group.
 | |||||
|---|---|---|---|---|---|
| Entry | Cmpd. | R1 | R2 | Time (h) | Yield (%)a) |
| 1 | 3a | 4-Nitrophenyl | C6H5 | 27 | 87 |
| 2 | 3b | 4-Methoxyphenyl | C6H5 | 44 | 75 |
| 3 | 3c | Benzyl | C6H5 | 96 | 0 |
| 4 | 3d | C6H5 | 4-Nitrophenyl | 20 | 91 |
| 5 | 3e | C6H5 | 4-Methoxyphenyl | 44 | 80 |
| 6 | 3f | C6H5 | Methyl | 97 | 2 |
a) Isolated yield.
The difference in the reactivity of various carbamates (3a–3d) in Table 2 led us to investigate selective formation of N,N′-diarylureas from a mixture of carbamates (Fig. 2); compatibility with multiple reactive sites would be beneficial for application to polymerization. A mixture of 3a and 3b was reacted under similar conditions to those of Table 2, affording a complex mixture of 4, 5 and other unidentified compounds (Fig. 2a). It is noteworthy that compound 6 was not observed in the crude mixture. At this stage, we think low nucleophilicity of p-nitroaniline generated from 3a might be a major reason that 6 was not observed. Similar reaction of 3d and 3g also resulted in a complex mixture of 2, 7, and 8 (ca. 2 : 7 : 8=1 : 2 : 1 based on the 1H-NMR of the crude mixture), together with other unidentified products (Fig. 2b). These results may imply that the reaction involves multiple steps.
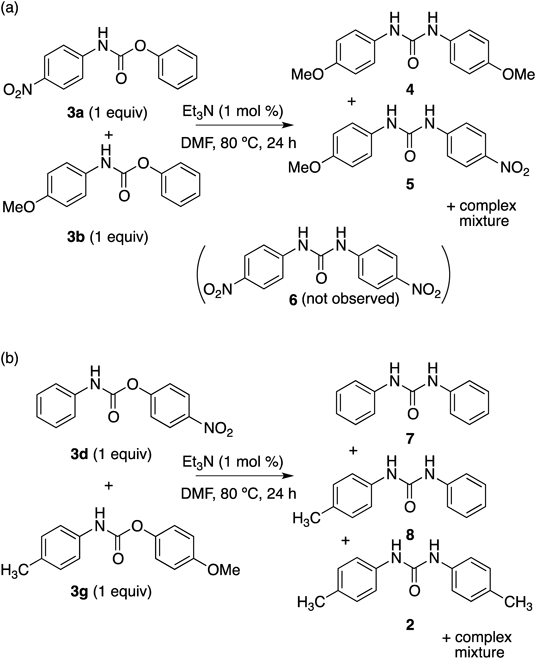
To obtain mechanistic insight into the reaction, the carbamate 3h was reacted with p-toluidine under the same conditions. An unsymmetrically substituted urea, 8 was obtained selectively in 90% yield within 1 h (Fig. 3a) as expected from literatures.22) The carbamate 3h was also reacted with p-tolyl isocyanate under the same conditions. Selectivity was low, and a complex mixture of 7, 8 and 2 (ca. 7 : 8 : 2=1 : 2 : 1 from 1H-NMR of the crude mixture) and other impurities was obtained (Fig. 3b). A comparison of reactivity in Table 2 and product ratios in Figs. 2b and 3b suggests that the product-determining step may be formation of aryl isocyanate, for which the presence of an electron-donating group would be disadvantageous.

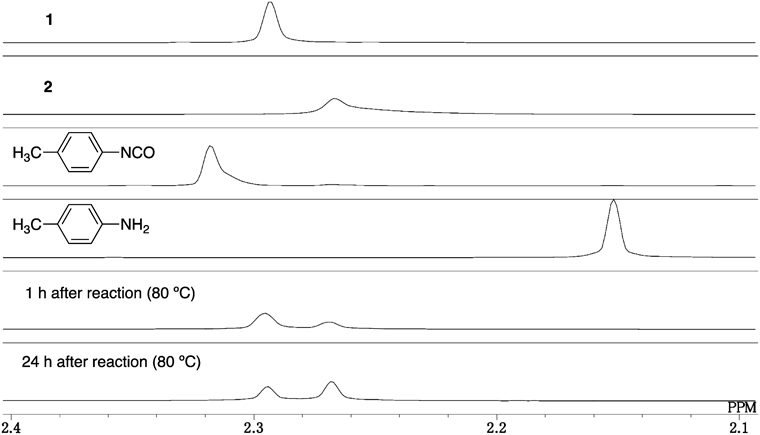
To observe intermediates involved in the reaction, we monitored the reaction of 1 in the presence of 1 mol % Et3N by means of 1H-NMR in DMF-d7 in a sealed tube at 80°C (Fig. 4). The reaction was slower, and carbamate was remained even after 24 h, probably due to the lack of moisture in a sealed tube. Signals of carbamate 1 and 2 were observed in the Ph-CH3 region (ca. 2 ppm), and those of p-tolyl isocyanate and p-toluidine were not detected. The absence of p-tolyl isocyanate and p-toluidine peaks in the reaction mixture suggests that the formation of aryl isocyanate and aniline from carbamate is slow (rate-limiting step) and/or that the equilibrium between carbamate and isocyanate predominantly favors carbamate,17) so that isocyanate is undetectable by 1H-NMR.
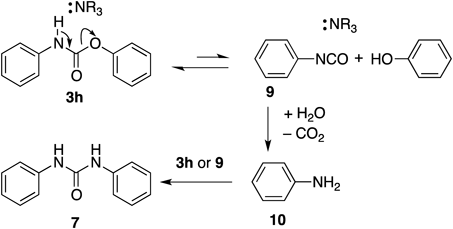
Based on the our findings and reported results,13,17) we propose the reaction mechanism depicted in Chart 1. NR3 (or Et3N) catalyzes the reaction of carbamate 3h to 9, which favors 3h in the equilibrium. Phenyl isocyanate 9 can be hydrolyzed by residual H2O to form aniline 10, which in turn reacts with 3h or 9 to afford N,N′-diphenylurea 7. Since absence of water, under a strict anhydrous condition, probably prevented the formation of 10, and the reaction was not completed (cf. Fig. 4).
We found that N,O-diaryl carbamate is spontaneously transformed into symmetrical N,N′-diphenylurea in DMF in the presence of a catalytic amount of Et3N. This transformation requires a presence of aryl groups on the nitrogen and oxygen atoms of the carbamate. An electron-donating group on the aryl group inhibited the reaction. We could not obtain selective coupling products from a mixture of carbamates, apparently due to involvement of multiple intermediates in the reaction. This reaction should be useful to synthesize functional molecules with an N,N′-diphenylurea skeleton via polymerization or cyclization by employing bench-stable N,O-diaryl carbamate.
Melting points (mp) were determined by using a Yanaco melting point apparatus MP-S3 and are uncorrected. Fourier transform (FT)-IR spectra were recorded on a Horiba FT-710. 1H- and 13C-NMR spectra were recorded on a JEOL AL-300 or ECZ400S or Bruker AV-300M, and chemical shifts are expressed in parts per million relative to tetramethylsilane (TMS) or residual solvent peak (7.24 ppm for CDCl3 (1H), 77.0 ppm for CHCl3 (13C), 2.49 ppm for DMSO-d6 (1H), 39.5 ppm for DMSO-d6 (13C), 8.03 ppm for DMF-d7 (1H)). Mass spectra were measured on a JEOL MS700V or HX110. Silica gel (CHROMATOREX PSQ100 (Fuji Silysia Chemical Co., Inc.)) was used for all chromatographic procedures.
Preparation of 1Et3N (0.80 mL, 5.7 mmol) and phenyl chloroformate (1.0 mL, 7.9 mmol) were added dropwise to a solution of p-toluidine (1.00 g, 9.3 mmol) in CH2Cl2 (10 mL) under cooling in an ice bath. The mixture was stirred for 1 h, and then evaporated to dryness. The residue was recrystallized from n-hexane to afford 1 (660 mg, 51%) as colorless needles. 1H-NMR and mp were consistent with reported data.23)
Phenyl N-(4-Methylphenyl)carbamate (1)mp 119.0–120.0°C; 1H-NMR (300 MHz, CDCl3) 2.33 (s, 3H), 7.13–7.24 (m, 5H), 7.32–7.42 (m, 4H).
General Procedure for the Reaction of 1 (Table 1)Et3N (1.4 µL, 0.01 mmol) was added to a solution of 1 (227 mg, 1.0 mmol) in DMF (4 mL), and the mixture was stirred at 80°C without Ar displacement (open-air) until the reaction was complete, as determined by TLC analysis. Then, Et2O (200 mL) was added, and the mixture was washed with sat. NH4Cl aq. (200 mL) and 10% NaOH aq. (200 mL) twice, dried over Na2SO4, and filtered. The filtrate was evaporated under reduced pressure to afford 2 as a colorless powder. 1H-NMR was consistent with reported data.24)
N,N′-Bis(4-methylphenyl)urea (2)1H-NMR (300 MHz, DMSO-d6) 2.22 (s, 6H), 7.06 (d, J=8.4 Hz, 4H), 7.31 (d, J=8.4 Hz, 4H), 8.49 (br s, 2H).
Preparation of 3aPyridine (0.77 mL, 9.6 mmol) and phenyl chloroformate (1.1 mL, 8.8 mmol) were added dropwise to a solution of p-nitroaniline (1.11 g, 8.0 mmol) in CH2Cl2 (50 mL), and the reaction mixture was stirred for 1 h at room temperature (r.t.). Then, 10% HCl (1 mL) and H2O (40 mL) were added. The organic layer was dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent, AcOEt–hexane=1 : 3) to afford 3a as a yellow solid (1.69 g, 81%). 1H-NMR and mp were consistent with reported data.25)
Phenyl N-(4-Nitrophenyl)carbamate (3a)mp 158.0–159.0°C; 1H-NMR (300 MHz, CDCl3) 7.20 (d, J=9.0 Hz, 2H), 7.31 (m, 2H), 7.40–7.45 (m, 2H), 7.62 (d, J=9.0 Hz, 2H), 8.24 (d, J=9.0 Hz, 2H).
Preparation of 3bCompound 3b was synthesized in a similar manner to that described for 3a, except that p-methoxyaniline was used in place of p-nitroaniline. It was purified by recrystallization from CH2Cl2 to afford colorless needles (71%). 1H-NMR and mp were consistent with reported data.25)
Phenyl N-(4-Methoxyphenyl)carbamate (3b)mp 150.5–151.0°C; 1H-NMR (300 MHz, CDCl3) 3.80 (s, 3H), 6.82 (br s, 1H), 6.88 (d, J=9.0 Hz, 2H), 7.18–7.23 (m, 3H), 7.35–7.42 (m, 4H).
Preparation of 3cA solution of phenyl chloroformate (1.2 mL, 9.5 mmol) in CH2Cl2 (5 mL) and tetrahydrofuran (THF) (5 mL) was added to a solution of benzylamine (1.0 mL, 9.15 mmol) and N,N-diisopropylethylamine (3.2 mL, 18.3 mmol) and in CH2Cl2 (10 mL) and THF (10 mL) in an ice bath. The mixture was stirred for 23 h at r.t., and then evaporated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent, AcOEt–hexane=1 : 7) to afford 3c as a colorless solid (1.31 g, 63%). 1H-NMR and mp were consistent with reported data.26)
Phenyl N-Benzylcarbamate (3c)mp 77.0–78.0°C; 1H-NMR (300 MHz, CDCl3) 4.47 (d, J=6.0 Hz, 2H), 5.33 (br s, 1H), 7.14–7.23 (m, 3H), 7.28–7.41 (m, 7H).
Preparation of 3dPhenyl isocyanate (0.8 mL, 7.3 mmol) was added to a solution of p-nitrophenol (1.00 g, 7.2 mmol) in CH2Cl2 (20 mL) in an ice bath. The reaction mixture was stirred for 19 h at r.t., and then evaporated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent, CH2Cl2–hexane=5 : 1) to afford 3d as a colorless solid (0.569 g, 31%). 1H-NMR and mp were consistent with reported values.27)
4-Nitrophenyl N-Phenylcarbamate (3d)mp 146.0–147.0°C; 1H-NMR (300 MHz, CDCl3) 6.91 (br s, 1H), 7.16 (t, J=7.2 Hz, 1H), 7.35–7.46 (m, 6H), 8.29 (d, J=9.0 Hz, 2H).
Preparation of 3eCompound 3e was synthesized in a similar manner to that described for 3d, except that p-methoxyphenol was used in place of p-nitrophenol. The product was purified by recrystallization from ethanol to afford a colorless solid (56%).
4-Methoxyphenyl N-Phenylcarbamate (3e)mp 138.0–139.0°C; 1H-NMR (300 MHz, CDCl3) 3.81 (s, 3H), 6.38–6.93 (m, 3H), 7.08–7.13 (m, 3H), 7.34 (t, J=7.5 Hz, 2H), 7.44 (d, J=7.5 Hz, 2H); 13C-NMR(75 MHz, CDCl3) 55.6, 114.4, 118.7, 122.5, 123.8, 129.1, 137.4, 144.1, 152.0, 157.2; IR (KBr) 3344, 2924 2856, 1722, 1606, 1549, 1502, 1448, 1381, 1317, 1213, 1022, 904, 839, 758, 696, 663, 519 cm−1; high resolution (HR)-MS (electron ionization (EI)+) Calcd for C14H13NO3 (M+) 243.0890. Found 243.0889.
Preparation of 3fPhenyl isocyanate (2.7 mL, 25 mmol) was added to dry methanol (40 mL) in a two-necked flask under Ar. The solution was stirred at 80°C for 30 min, and evaporated under reduced pressure. The residue was cooled in a dry ice-acetone bath, affording a colorless solid 3f (3.67 g, 98%). 1H-NMR and mp were consistent with reported data.28)
Methyl Phenylcarbamate (3f)mp 43.0–44.0°C; 1H-NMR (300 MHz, CDCl3) 3.78 (s, 3H), 6.61 (br s, 1H), 7.07 (t, J=7.5 Hz, 1H), 7.31 (t, J=7.5 Hz, 2H), 7.38 (t, J=7.5 Hz, 2H).
Preparation of 3gEt3N (0.8 mL, 5.7 mmol) and p-tolyl isocyanate (1.5 mL, 12 mmol) were added to a solution of p-methoxyphenol (1.50 g, 12 mmol) in toluene (57 mL) at r.t. The reaction mixture was stirred for 3 h at r.t., and then evaporated under reduced pressure. The crude product was recrystallized from ethanol to afford 3g as colorless needles (2.18g, 71%). 1H-NMR and mp were consistent with reported data.29,30)
4-Methoxyphenyl N-(4-Methylphenyl)carbamate (3g)mp 164.5–165.0°C; 1H-NMR (300 MHz, CDCl3) 2.32 (s, 3H), 3.80 (s, 3H), 6.90 (d, J=9.0 Hz, 2H), 7.08–7.14 (m, 4H), 7.32 (m, 2H).
Preparation of 3hCompound 3h was synthesized in a similar manner to that described for 1, except that aniline was used in place of p-toluidine. The product was purified by silica gel column chromatography (eluent, AcOEt–hexane=1 : 3), then recrystallization from CH2Cl2 to afford 3h as colorless prisms (19%). 1H-NMR and mp were consistent with reported data.23)
Phenyl N-Phenylcarbamate (3h)mp 130.0–131.0°C; 1H-NMR (300 MHz, CDCl3) 6.91 (br s, 1H), 7.11 (t, J=7.5 Hz, 1H), 7.18–7.47 (m, 9H).
Compound Data Related to Table 2N,N′-Bis(4-methoxyphenyl)urea (4)1H-NMR was consistent with reported data31); 1H-NMR (300 MHz, DMSO-d6) 3.70 (s, 6H), 6.84 (d, J=8.1 Hz, 4H), 7.32 (d, J=8.1 Hz, 4H), 8.36 (br s, 2H).
N,N′-Bis(4-nitrophenyl)urea (6)1H-NMR was consistent with reported data32); 1H-NMR (300 MHz, DMSO-d6) 7.72 (d, J=9.1 Hz, 4H), 8.22 (d, J=9.1 Hz, 4H), 9.65 (br s, 2H).
N-Phenyl-N′-(4-methylphenyl)urea (8)1H-NMR was consistent with reported data33); 1H-NMR (300 MHz, DMSO-d6) 2.23 (s, 3H), 6.94 (t, J=7.4 Hz, 1H), 7.07 (d, J=8.1 Hz, 2H), 7.26 (t, J=8.1 Hz, 2H), 7.32 (d, J=8.4 Hz, 2H), 7.43 (d, J=8.1 Hz, 2H), 8.52 (br s, 1H), 8.59 (br s, 1H).
Preparation of 5Synthesized for the purpose of comparison in the 1H-NMR experiment in Fig. 2a. A solution of p-anisidine (246 mg, 2.0 mmol) in Et2O (2 mL) was added to a solution of p-nitrophenyl isocyanate (328 mg, 2.0 mmol) in Et2O (5 mL) in an ice bath. The mixture was stirred for 16 h at r.t., then the precipitate was collected by suction, and washed with Et2O. The crude product was recrystallized from methanol to afford 5 as a colorless powder (134 mg, 24%).
N-(4-Methoxyphenyl)-N′-(4-nitrophenyl)urea (5)mp 236.5–238.0°C; 1H-NMR (400 MHz, DMSO-d6) 3.71 (s, 3H), 6.88 (d, J=9.2 Hz, 2H), 7.37 (d, J=9.2 Hz, 2H), 7.67 (d, J=9.2 Hz, 2H), 8.17 (d, J=8.1 Hz, 2H), 8.73 (br s, 1H), 9.37 (br s, 1H); 13C-NMR (100 MHz, DMSO-d6) 55.2, 114.0, 117.3, 120.5, 125.2, 131.9, 140.8, 146.6, 152.1, 154.9; IR (KBr) 2954, 1691, 1645, 1614, 1593, 1552, 1506, 1468, 1406, 1342, 1304, 1228, 1167, 1109, 1032, 858, 761, 744, 688, 548 cm−1; HR-MS (EI+) Calcd for C14H13N3O4 (M+) 287.0901. Found 287.0906.
Reaction Monitoring by 1H-NMR (Fig. 4)A mixture of 1 (42.6 mg, 0.187 mmol) and Et3N (2.6 µL, 1.9 µmol) in DMF-d7 (0.675 mL) was heated in an NMR tube in an oil bath at 80°C. The 1H-NMR spectrum of the mixture was measured at the indicated times.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials. Detail and 1H-NMR spectrums of competitive reaction in Figs. 2 and 3; 1H- and 13C-NMR spectrums of compound 3e.