Review
Development of Chemical Biology Tools Focusing on Peptide/Amide Bond Cleavage Reaction
2019 Volume 67 Issue 11 Pages 1171-1178
Details
2019 Volume 67 Issue 11 Pages 1171-1178
Peptides and proteins are involved in almost all biological events. In this review, three chemical biology tools, which were developed for peptide/protein sciences from a viewpoint of peptide/amide bond cleavage, are overviewed. First, study on an artificial amino acid that enables stimulus-responsive functional control of peptides/proteins is briefly described. Two N–S acyl transfer reaction-based tools, one a linker molecule for facile identification of target proteins of bioactive compounds and the other a reagent for selective labeling of proteins of interest, are then discussed.
Peptides and proteins are key players of almost all biological events. For elucidation of functions of endogenous peptides/proteins, they or their derivatives with an artificial moiety such as a fluorophore are widely employed. To accelerate these researches, many groups have focused on chemical peptide/protein synthesis because it enables facile access and continuous supply of naturally occurring and artificial peptides/proteins. Because a main theme of peptide/protein chemistry is “formation of peptide bond,” many researchers had already flooded this field when my academic career began in 2005. I thought that “formation of peptide bond” is Red Ocean, but “cleavage of peptide/amide bond” might be Blue Ocean.1) I therefore decided to invent peptide/amide bond cleavage-based chemical biology tools for the peptide/protein sciences.
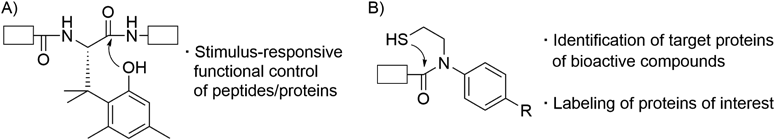
In this review, our three chemical biology tools based on peptide/amide bond cleavage are described. First, study on an artificial amino acid that enables stimulus-responsive functional control of peptides/proteins is overviewed (Fig. 1A). Two N–S acyl transfer reaction-based tools, one a linker molecule for facile identification of target proteins of bioactive compounds and the other a reagent for selective labeling of proteins of interest, are then discussed (Fig. 1B).
Because details of the stimulus-responsive amino acid were reported in our recent review,2) it is only briefly described in this section.
Artificial peptides and proteins whose function can be controlled by external stimuli are indispensable tools in the field of peptide/protein sciences. A typical example is photo-responsive caged peptides and proteins.3,4) Several caged peptides and proteins, which undergo UV-responsive main chain cleavage to alter their bioactivities, were developed to clarify mechanisms of action of original uncaged peptides/proteins.5–8) These caged peptides/proteins contain a UV-responsive artificial amino acid such as 2-nitrophenylglycine; therefore laborious de novo design and synthesis of a new amino acid will be required if the peptides/proteins should respond to a stimulus other than UV. In this context, we started to invent a stimulus-responsive peptide-bond-cleaving residue (Spr), which can respond to various stimuli by just replacing its protecting group2) (Fig. 1A).
Because we wanted to use a Spr-containing peptide/protein in living cells, Spr should induce peptide bond cleavage under physiological conditions. However, it is well known that chemical peptide/amide bond cleavage requires harsh conditions. We therefore searched amide bond cleavage reaction that occurs under mild conditions, and a trimethyl lock system depicted in Fig. 2A was fortunately found.9–12) Trimethyl lock derivative 1 (X = NH) induces amide bond cleavage even under mild conditions by assistance of steric repulsion between three methyl groups depicted as Me. Inspired by the trimethyl lock system, we designed Spr shown in Fig. 2B. Spr is an amino acid derivative of the trimethyl lock unit. It possesses a protecting group (PG) on the phenolic hydroxyl group; removal of the PG induces peptide bond cleavage. In this case, replacement of the PG should enable Spr to respond to various stimuli.
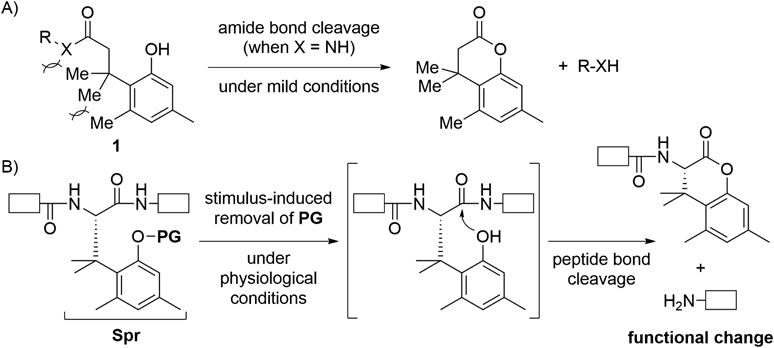
A) Trimethyl lock system; B) Stimulus-responsive peptide bond cleavage by Spr. PG: protecting group; Spr: stimulus-responsive peptide-bond-cleaving residue; white boxes: peptide.
Details of synthesis of Spr and Spr-containing peptides are omitted in this review, but Spr derivatives for solid phase peptide synthesis (SPPS) were enantioselectively prepared13) and could be incorporated into peptides by standard Fmoc-based and Boc-based SPPS.14) Structures of PG unit of Spr developed so far and corresponding stimuli are summarized in Fig. 3. Not only UV13,14) but also near-IR (NIR) two-photon excitation,15) intracellular hypoxic environment,16) thiol,17,18) hydrogen peroxide,19) fluoride anion,20) and alkaline phosphatase14) can trigger peptide bond cleavage of Spr. Several Spr-containing peptides were already used to control function of peptides/proteins in living cells.14,16,21) Spr has been recently employed by some other groups for in-cell imaging of a protein of interest,21) UV-induced propulsion of a giant liposome,22) UV-triggered nanofiber formation,23) and tumor-targeting therapy.24) Application of Spr in topics not mentioned in this paper14,17,18,25,26) are detailed in our recent review.2)
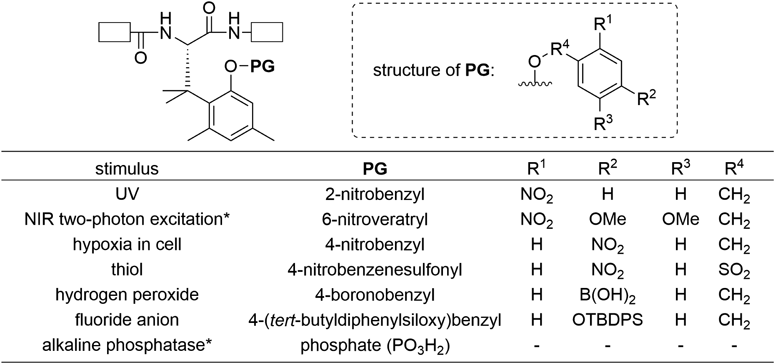
NIR: near-IR; white boxes: Peptide; *: Racemic Spr was prepared.
We have been developing new methodologies for chemical protein synthesis. A reaction commonly used in this field is native chemical ligation27–29) (NCL, Fig. 4). It enables chemoselective condensation of a peptide fragment with a C-terminal thioester and an N-cysteinyl peptide fragment via intermolecular S–S acyl transfer followed by intramolecular S–N acyl transfer to generate a ligated peptide or protein under mild aqueous conditions. To synthesize large proteins, condensation of more than two peptide fragments is usually required.30) In this case, temporal protection of the C-terminal thioester or the N-terminal cysteine is sometimes needed to realize sequential NCL.
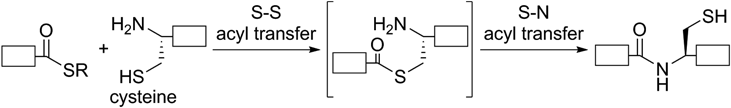
White box: peptide.
In this context, we previously developed N-sulfanylethylanilide (SEAlide) for the protection and on-demand generation of the C-terminal thioester31–33) (Fig. 5A). SEAlide is stable in the absence of phosphate; however, it can be converted to a thioester form by addition of phosphate to enable subsequent reaction with nucleophiles.34,35) Plausible mechanism of the phosphate-catalyzed generation of the thioester is depicted in Fig. 5B.34,36) Taking pKas of phosphate into account, it could work as an acid-base catalyst to accelerate generation of the thioester by protonation and deprotonation of SEAlide. This implies that SEAlide can be regarded as an auxiliary that induces acid-base catalyst-responsive metathesis of R2–R1 (2) to R2–R3 (3) via amide bond cleavage followed by new bond formation (Fig. 5C). We thought that the conditional metathesis of SEAlide could be applied to new tools for peptide/protein sciences. SEAlide-based tools for identification of target proteins of bioactive compounds and selective labeling of proteins of interest are overviewed below.

A) Phosphate-catalyzed thioester formation of SEAlide; B) Plausible mechanism of the thioester formation catalyzed by phosphate as acid-base catalyst; C) Acid-base catalyst-responsive metathesis of the SEAlide derivative from R2–R1 (2) to R2–R3 (3). SEAlide: N-sulfanylethylanilide.
Elucidation of mechanism of action of bioactive ligands is key issue of drug development, and enrichment of target proteins of the ligands is indispensable for their identification. A cleavable linker is widely used for the enrichment; details are shown in Fig. 6A.37–39) A ligand derivative, which possesses an alkyne moiety and a crosslinking group such as a photo-affinity group,40–44) is first selectively introduced onto the target protein by covalent manner (step 1). Then, the cleavable linker with an azide and biotin units are conjugated with the alkyne-containing target by copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) (step 2).45–47) Following selective adsorption of the biotinylated target protein on streptavidin (SAv) beads48,49) and subsequent washing to remove non-targets (step 3), cleavage of the linker enables elution of the target protein without harsh conditions such as thermal denaturation of the SAv in the presence of sodium dodecyl sulfate (SDS) (step 4). The methodology employing the cleavable linker is useful and practical; however, contamination of the non-targets derived from non-specific adsorption is sometimes observed and this may hamper subsequent target identification.50–52)
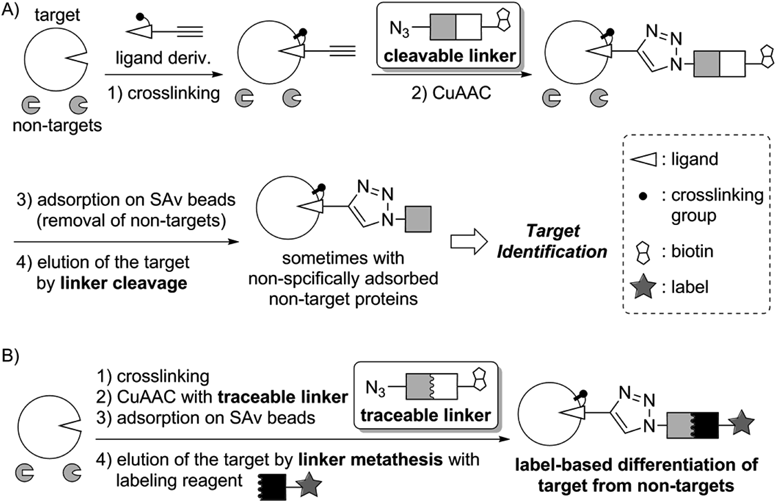
In this context, a new linker that enables differentiation of the target protein from non-targets was developed18,20,53–56) (Fig. 6B). Steps 1 to 3 are similar to those of the cleavable linker. In step 4, metathesis of the biotin unit with a labeling reagent is conducted as follows. Cleavage of the linker generates bio-orthogonal functional groups, which are then selectively labeled to differentiate the target protein from non-targets. Because this linker enables label-based tracing of the target, we coined the linker “traceable linker.”18) The traceable linker reported so far uses reversible reaction to label the target (e.g. oxime or hydrazone formation); therefore the label moiety would be accidentally removed in the subsequent manipulation. Hence we decided to develop an irreversible reaction-based traceable linker.
A SEAlide-based traceable linker is shown in Fig. 7.56) The linker is stable until addition of phosphate buffer at the linker metathesis step. Once the phosphate buffer and a cysteine-label conjugate are added, activation of the SEAlide moiety to a thioester form followed by irreversible NCL with the cysteine derivative should induce linker cleavage and selective labeling of the target protein.

After optimization of the spacer moieties and enrichment conditions, a proof-of-concept experiment was performed. Alkynylated bovine serum albumin (alk-BSA) was mixed in a cell lysate (Fig. 8. Lane 1), and enrichment and selective labeling of alk-BSA using the SEAlide-based traceable linker was tested. In this experiment, a fluorescein–cysteine conjugate was employed for the linker cleavage and labeling. After enrichment, three proteins including alk-BSA were detected by silver staining (Fig. 8, Lane 2). Even though we would not distinguish alk-BSA from contaminated proteins if a cleavable linker was employed, the traceable linker successfully indicated alk-BSA by visualizing the label as we designed (Fig. 8, Lane 3). Application of the SEAlide-based traceable linker for identification of unknown target proteins of bioactive compounds is in progress.
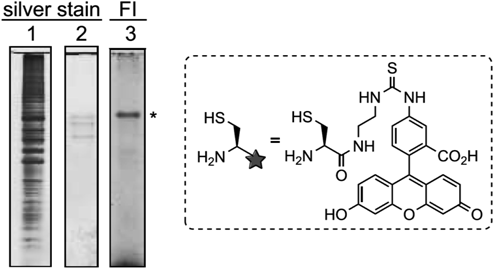
Lane 1: a mixture of alk-BSA and HCT116 cell lysate before enrichment; Lanes 2 and 3: eluted proteins after enrichment using the SEAlide-based traceable linker. Experimental details are described in reference 56. FI: fluorescence imaging; *: alk-BSA. (Adapted from reference 56 by permission of The Royal Society of Chemistry.)
Labeled and chemically modified proteins are in increasingly widespread use to study the structure, function, and localization of proteins.57–59) The labeling and chemical-modifying technology, which are recently attracting much attention, are affinity-based labeling such as represented by ligand-directed tosyl chemistry developed by Hamachi and colleagues.60–62) General design of this type of protein-labeling reagent is shown in Fig. 9A. The labeling reagent comprises a ligand and a label connected by electrophilic reactive unit. That can be bound on the target protein by ligand docking, and then a nucleophilic side chain near to the labeling reagent reacts with its reactive unit to generate a labeled target protein even in the presence of non-target proteins. Although these labeling reagents can successfully work under optimized conditions, a potential risk of side reactions such as hydrolysis of the reagents and labeling of non-targets are a concern because of their intrinsic reactivity.63) We thought that a labeling reagent, which is stable in the absence of its target protein but can be activated to a reactive form by binding to the target, should allow for target labeling without side reactions. It is well known that polar amino acids including acidic and basic residues are preferentially found on the surface of proteins. Taking this observation into account, a SEAlide-based Labeling reagent (SEAL) shown in Fig. 9B was designed.64) SEAL is stable in the absence of the target; however, it is activated to a thioester form by binding to the target because acidic and basic residues on the protein surface near to the binding site could work as an acid-base catalyst. The thioester can react with proximal nucleophilic residues; therefore target-activated labeling is achieved.

A) General affinity-based labeling reagent; B) SEAlide-based labeling reagent (SEAL).
We first examined labeling of human carbonic anhydrase 1 (hCA1) in protein mixture (Fig. 10, Entry 1). By the use of SEAL with fluorescein and benzenesulfonamide, which is a ligand of hCA1, selective labeling of hCA1 in a protein mixture was observed. The generality of the SEAL technology was then confirmed by replacing the label and ligand units (Fig. 10, Entry 2). In this experiment, biotin-labeling of cyclooxygenase 1 (COX-1) in a protein mixture was successfully achieved. An S-methylated SEAL derivative did not label the target protein; therefore it was suggested that the labeling occurred via activation of SEAL to the thioester form. Furthermore, we speculated that the SEAL was activated by target protein because SEAL was intact in the presence of a high concentration of nucleophiles.65)
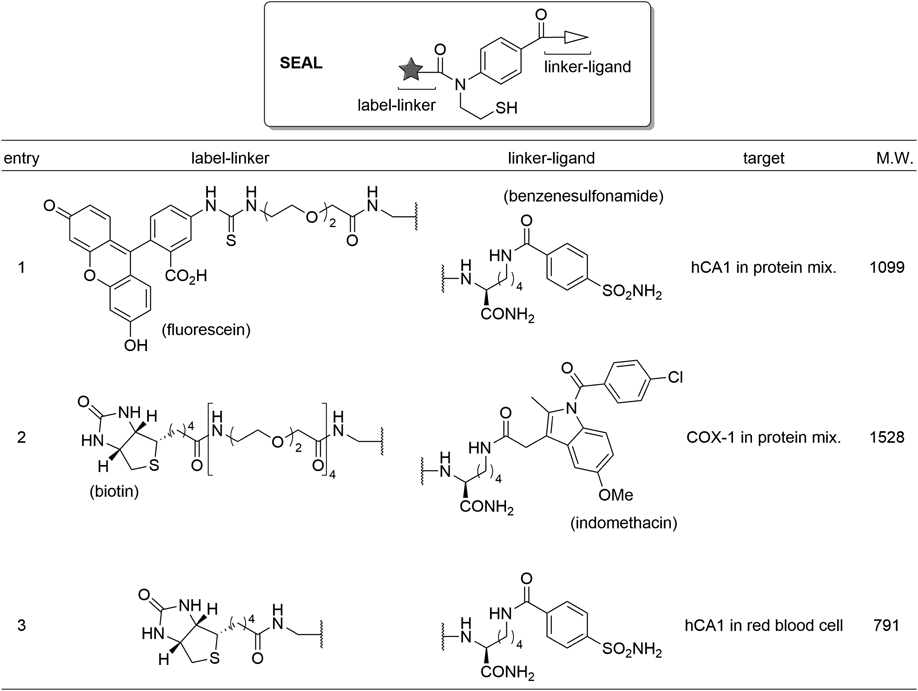
COX-1: cyclooxygenase 1; hCA1: human carbonic anhydrase 1; M.W.: molecular weight.
Encouraged by these results, we applied the SEAL technology to (1) in cell labeling of the target proteins and (2) elucidation of a ligand-binding pocket of the target protein.
3.3.2. Application of SEAL to in Cell LabelingTo elucidate the functions of proteins, studies not only in a test tube but also in cells are invaluable. Affinity-based labeling is an exciting emerging methodology in this field because it enables introduction of a small label onto a target endogenous protein in living cells.59–61) We therefore challenged to apply SEAL to in cell labeling.64) SEAL developed to label hCA1 in protein mixture was first employed to label hCA1 in red blood cells (Fig. 10, Entry 1). However, the labeled hCA1 was not detected probably due to low cell permeability of the SEAL. Reduction of molecular weight and removal of negative charge was therefore attempted, and the new SEAL successfully enabled in cell labeling of hCA1 in red blood cells (Fig. 10, Entry 3). Since only one example of the in cell labeling by SEAL has been demonstrated, it is necessary to verify its generality and improve efficacy of the labeling in future.
3.3.3. Elucidation of Ligand-Binding Pocket Using SEALIt is known that amino acids near to a protein’s ligand-binding pocket are preferentially labeled by ligand-directed labeling.60) Therefore the position of labeled residue gives us an insight into location of the binding pocket. SEAL was also confirmed to label a lysine (Lys) residue proximal to the pocket.64) To demonstrate applicability of SEAL to elucidate an unknown binding pocket, we decided to uncover an inhibitor-binding pocket of human D-amino acid oxidase (hDAO).66) An inhibitor of hDAO is expected as new therapeutics for schizophrenia67); therefore screening of the inhibitors was conducted by Fukui et al. and they found 4-bromo-3-nitrobenzoic acid (BNBA, Fig. 11A) as a hit compound. Collaborating with Fukui’s group, we planned to clarify a BNBA-binding pocket of hDAO by combining SEAL technology and in silico docking study. First, the docking study of BNBA to hDAO dimer was examined and two pockets were predicted as shown in Fig. 11C (depicted as predicted binding site A and B). The site A is reasonable because there is an entrance of a substrate-binding pocket, in which inhibitors such as benzoate are known to bind68); however, the site B was unexpected. The predicted pockets were then verified using the SEAL technology. By the use of SEAL possessing a BNBA analog as the ligand unit and biotin as the label unit (Fig. 11B), labeling at Lys163 and Lys204 was observed. Both Lys residues are near to the computationally predicted pockets; therefore the presence of two binding pockets, one of which was unexpected, was demonstrated by combining in silico docking study and SEAL technology. Clarification whether binding of BNBA to the site B is responsible for the inhibition of hDAO is a challenge for the future.69,70)
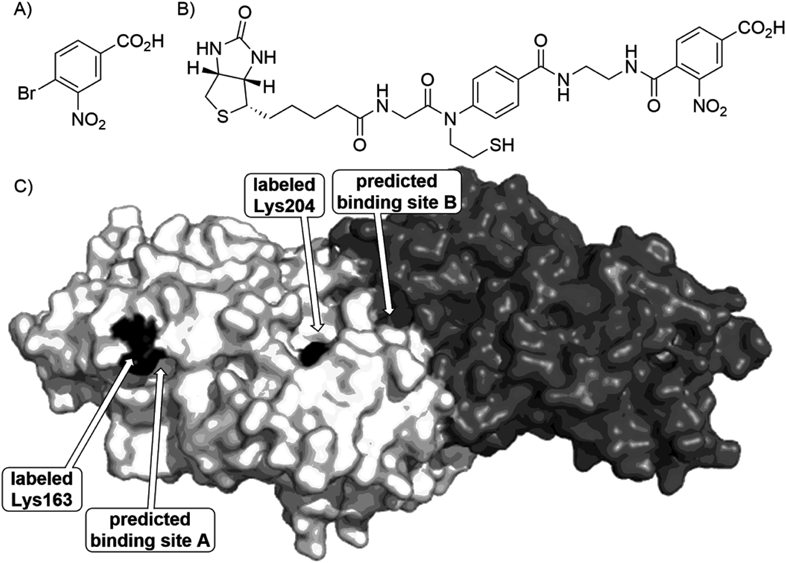
A) Structure of 4-bromo-3-nitrobenzoic acid (BNBA); B) Structure of SEAL with a BNBA-derived ligand moiety; C) Labeled residues and computationally predicted BNBA-binding pockets of hDAO dimer (PDB ID: 2du868)).
Focusing on peptide/amide bond cleavage reaction, three chemical biology tools were overviewed: 1) Spr that is an artificial amino acid for stimulus-responsive functional control of peptides and proteins2); 2) SEAlide-based traceable linker as an advanced cleavable linker for enrichment of target proteins of bioactive compounds56); 3) SEAL for protein labeling that is applicable to in cell labeling64) and clarification of a ligand-binding pocket of proteins.66) Next step of these studies will be validation of their practicality; therefore application of these tools to answer biological and medicinal questions such as elucidation of unknown target proteins of bioactive ligands is in progress. I hope that our tools developed from chemical point of view will contribute to expansion of biomedical knowledge and development of novel therapeutics in future.
The author expresses sincere appreciation to Professor Akira Otaka (Tokushima University) for his generous guidance and continuous encouragement. The author is also grateful to his collaborators especially Dr. Jun Yamamoto, Professor Masaya Denda (Kyoto University), Dr. Takuya Morisaki, Mr. Taiki Kohiki, Professor Kohei Sato (Shizuoka University), and Professor Tsubasa Inokuma (Tokushima University) for their critical contributions to the research reviewed in this paper. This body of research was supported in part by PRESTO, Japan Science and Technology Agency (JST, Grant Number JPMJPR1333), Grant-in-Aid for Scientific Research (KAKENHI, Grant Number 15K07858, 22790012, 20790012), Ajinomoto Co., Inc. (Ajinomoto Award in Synthetic Organic Chemistry, Japan), Mitsubishi Chemical Corporation Fund, The Takeda Science Foundation, The Japan Prize Foundation, Astellas Foundation for Research on Metabolic Disorders, Takeda Pharmaceutical Company Limited (Takeda Pharmaceutical Company Award in Synthetic Organic Chemistry, Japan), and The Research Foundation for Pharmaceutical Sciences.
The author declares no conflict of interest.