
- |<
- <
- 1
- >
- >|
-
 Akira Shigenaga2019Volume 67Issue 11 Pages 1171-1178
Akira Shigenaga2019Volume 67Issue 11 Pages 1171-1178
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
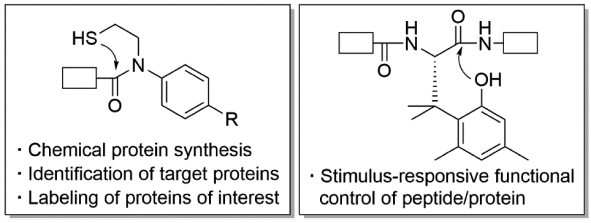
JOURNAL FREE ACCESS FULL-TEXT HTMLPeptides and proteins are involved in almost all biological events. In this review, three chemical biology tools, which were developed for peptide/protein sciences from a viewpoint of peptide/amide bond cleavage, are overviewed. First, study on an artificial amino acid that enables stimulus-responsive functional control of peptides/proteins is briefly described. Two N–S acyl transfer reaction-based tools, one a linker molecule for facile identification of target proteins of bioactive compounds and the other a reagent for selective labeling of proteins of interest, are then discussed.
Graphical Abstract Fullsize ImageView full abstractEditor's pickElucidation of functions of endogenous peptides/proteins is undoubtedly valuable because they are key players of most of biological pathways. The author has developed chemical biology tools to accelerate the functional elucidation from the viewpoint of synthetic organic chemistry. In this Featured Article, the author introduces following three tools invented by his group: 1) an artificial amino acid that enables stimulus-responsive functional control of peptides and proteins; 2) a traceable linker for facile identification of target proteins of bioactive ligands; 3) an in-cell compatible labeling reagent of proteins of interest.
Download PDF (1672K) Full view HTML
-
Deprotonative Coupling of Pyridines with Aldehydes Catalyzed by an HMDS-Amide Base Generated in SituMasanori Shigeno, Kunihito Nakaji, Akihisa Kajima, Kanako Nozawa-Kumad ...2019Volume 67Issue 11 Pages 1179-1182
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialHerein, the deprotonative functionalization of pyridine derivatives with aldehydes under ambient conditions has been demonstrated using an amide base generated in situ from a catalytic amount of CsF and a stoichiometric amount of tris(trimethylsilyl)amine (N(TMS)3). Pyridine substrates bearing two electron-withdrawing substituents (i.e., fluoro, chloro, bromo, and trifluoromethyl moieties) at the 3- and 5-positions efficiently react at the 4-position with various aldehydes including arylaldehydes, pivalaldehyde, and cyclohexanecarboxaldehyde.
Graphical Abstract Fullsize ImageView full abstractEditor's pickDeprotonative coupling of pyridines with electrophiles using a stoichiometric amount of strong Brønsted bases usually employs cryogenic conditions in order to inhibit the occurrence of side-reactions. In this paper, the authors established the efficient and convenient coupling reaction of 3,5-dihalopyridines and 3-chloro-5-(trifluoromethyl)pyridine using an HMDS-amide base generated in situ from a catalytic amount of CsF and a stoichiometric amount of N(TMS)3. The reaction proceeds under ambient conditions and demonstrates the applicability of various (hetero)arylaldehydes, pivalaldehyde, and cyclohexanecarboxaldehyde as an electrophile.
Download PDF (497K) Full view HTML
-
Atsuko Sato, Naoki Tanimura, Teruki Honma, Akihiko Konagaya2019Volume 67Issue 11 Pages 1183-1190
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
Advance online publication: August 17, 2019 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialFor rational drug design, it is essential to predict the binding mode of protein–ligand complexes. Although various machine learning-based models have been reported that use convolutional neural networks (deep learning) to predict binding modes from three-dimensional structures, there are few detailed reports on how best to construct and use datasets. Here, we examined how different datasets affected the prediction of the binding mode of CYP3A4 by a three-dimensional neural network when the number of crystal structures for the target protein was limited. We used four different training datasets: one large, general dataset containing various protein complexes and three smaller, more specific datasets containing complexes with CYP3A4-like pockets, complexes with CYP3A4-binding ligands, and complexes with CYP protein family members. We then trained models with different combinations of datasets with or without subsequent fine-tuning and evaluated the binding mode prediction performance of each model. The best receiver operating characteristic (ROC) area under the curve (AUC) model with respect to area under the receiver operating characteristic curve was obtained by training with a combination of the general protein and CYP family datasets. However, the ROC AUC—recall balanced model was obtained by training with this combination of datasets followed by fine-tuning with the CYP3A4-binding ligands dataset. Our results suggest that datasets that balance protein functionality and data size are important for optimizing binding mode prediction performance. In addition, datasets with large median binding pocket sizes may be important for the binding mode prediction specifically of CYP3A4.
Graphical Abstract Fullsize ImageView full abstractEditor's pickA data science methodology to identify the correct binding mode between CYP3A4 and compounds using deep learning has been developed. The methodology enables us to predict the biding mode between a substrate and CYP3A4 whose binding mode is difficult to predict with conventional docking algorithms and binding mode scores mainly due to the larger and more flexible biding pocket than that of the other CYPs.
Download PDF (1934K) Full view HTML -
Shafaq Mubarak, Muhammad Zia-Ur-Rehman, Nadia Jamil, Muhammad Zaheer, ...2019Volume 67Issue 11 Pages 1191-1200
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialAn environment friendly synthesis of novel hybrid pharmacophores derived from synergism of nalidixic acid and 1,3-diphenylprop-2-en-1-ones is described. Percent yield and reaction times of microwave assisted reactions have been compared with the reactions carried out under conventional reaction conditions which show marked decrease in reaction times and significant increase in yields. Besides, anti-oxidant potential of the synthesized hybrid compounds was evaluated and some of the compounds showed marked ascorbic acid equivalence Ferric reducing anti-oxidant power (FRAP) and metal chelating capacities. Crystal study of one representative of the synthesized series is also presented.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (847K) Full view HTML -
Weijia Li, Fan Yang, Lingkuan Meng, Jiaqi Sun, Yangqing Su, Liang Shao ...2019Volume 67Issue 11 Pages 1201-1207
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
Advance online publication: August 22, 2019 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialOleanolic acid (OA) was discovered as a mild influenza hemagglutinin (HA) inhibitor in our earlier studies. In the present work, 20 compounds were prepared by structural modifications of OA, and their antiviral activities against influenza A/WSN/33 (H1N1) virus in Madin–Darby canine kidney (MDCK) cells were evaluated. Based on the biological result, structure–activity relationship (SAR) was discussed. Compound 10 with six-carbon chain and a terminal hydroxyl group showed the strongest anti-influenza activity with an IC50 of 2.98 µM, which is an order of magnitude more potent than OA. Hemagglutination inhibition and Surface plasmon resonance (SPR) assay indicated that compound 10 might interfere with influenza invasion by interacting with HA protein.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (1346K) Full view HTML -
Ziad Omran, Mohamed Alarja, Ashraf N. Abdalla, Munjed M. Ibrahim, Moha ...2019Volume 67Issue 11 Pages 1208-1210
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
Advance online publication: September 06, 2019 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialCo-drug, or mutual-prodrug, is a drug design approach consisting of covalently linking two active drugs so as to improve the pharmacokinetics and/or pharmacodynamics properties of one or both drugs. Co-drug strategy has proven good success in overcoming undesirable properties such as absorption, poor bioavailability, nonspecificity, and gastrointestine tract (GIT) side effects. In this work, we successfully developed a co-drug of 14-hydroxytylophorine, a phenanthroindolizidine derivative with remarkable antiproliferative activity, and dichloroacetate, a known inhibitor of pyruvate dehydrogenase kinase. Dichloroacetate steers tumour cell metabolism from glycolysis back to glucose oxidation, which in turn reverses the Warburg effect and renders tumour cells with a proliferative disadvantage. The obtained co-drugs retained the cytotoxicity of 14-hydroxytylophorine. However, they showed similar unselectivity towards normal cells.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (434K) Full view HTML -
Ko Morishita, Tomohiro Miike, Shigemitsu Takeda, Masaki Fukui, Yuma It ...2019Volume 67Issue 11 Pages 1211-1224
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLA novel series of 2,6,7-substituted 3-unsubstituted 1,2,3,4-tetrahydroisoquinoline derivatives were synthesized to find a peroxisome proliferator-activated receptor γ (PPARγ) partial agonist. Among the derivatives, (E)-7-[2-(cyclopent-3-eny)-5-methyloxazol-4-ylmethoxy]-2-[3-(2-furyl)acryloyl]-6-(1H-tetrazol-5-yl)-1,2,3,4-tetrahydroisoquinoline (20g) exhibited potent partial agonist activity (EC50 = 13 nM, maximal response 30%) and very weak protein tyrosine phosphatase 1B (PTP1B) inhibition (IC50 = 1100 nM), indicating a selective PPARγ partial agonist. A computational docking calculation revealed that 20g bound to PPARγ in a similar manner to that of known partial agonists. In male and female KK-Ay mice with insulin resistance and hyperglycemia, 20g at 30 mg/kg for 7 d significantly reduced plasma glucose levels, but not triglyceride levels. The effects of 20g were similar to those of pioglitazone at 10 mg/kg. In conclusion, the 2,6,7-substituted 1,2,3,4-tetrahydroisoquinoline with an acidic group at the 6-position provides a novel scaffold for selective PPARγ partial agonists and 20g exerted anti-diabetic effects via the partial activation of PPARγ.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (3523K) Full view HTML -
Mitsunobu Yoshida, Shinya Uchida, Yasuharu Kashiwagura, Shimako Tanaka ...2019Volume 67Issue 11 Pages 1225-1231
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLSolifenacin (Sol), an antimuscarinic agent has been widely used for the treatment of overactive bladder. Transdermal formulations can be administered without water as well as absorbed slowly into the blood over a long period of time. The aim of this study was to develop cream and tape formulations of Sol, and evaluate the transdermal permeation and absorption of the drug from the two formulations in vitro and in vivo, respectively. In the preparation of cream formulation, Sol succinate was dissolved in purified water, and the mixture was added to the hydrophilic cream. Then, aqueous sodium hydroxide was added to the cream. In the tape formulation, Sol succinate was dissolved in a solvent with propylene glycol, diisopropanolamine, triethyl citrate, and EUDRAGIT E100. The dissolved solvent was poured onto a polyethylene film. Cream (5%) and tape (15%) formulations demonstrated high skin permeability. Addition of an adsorption enhancer (N-methyl-2-pyrrolidone) did not further increase the level of skin permeability. In subsequent in vivo experiments in rats, both the cream and tape formulations led to slow absorption of Sol into plasma, with increased t1/2 compared with oral administration. Plasma Sol concentrations peaked 24 h after transdermal application and the drug was still detectable in plasma 72 h after application. Additionally, the cream (5%) and tape (15%) formulations resulted in a higher area under the plasma concentration vs. time curve from 0 to 72 h (AUC0–72) compared with oral formulation (30 mg/kg). In conclusion, significant in vitro permeability and in vivo absorption of Sol from the transdermal formulations were observed.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (511K) Full view HTML -
Lihong Yu, Liandi Chen, Guolin Luo, Licai Liu, Wenqi Zhu, Pengke Yan, ...2019Volume 67Issue 11 Pages 1232-1241
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
Advance online publication: September 06, 2019 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialTyrosinase plays a key role in the melanin biosynthesis since it catalyzes the transformation of tyrosine into L-dopaquinone. A large number of studies have also shown that molecules to efficiently inhibit the activity of tyrosinase would be potentially used in treating many depigmentation-related disorders. In this study, we targeted a series of structure-based 3-aryl substituted xanthone derivatives in which diverse functional groups were respectively attached on 3-aromatic ring moiety as new tyrosinase inhibitors. The results demonstrated that all obtained compounds had potent tyrosinase inhibitory activities with IC50 values at micromolar range. Especially, compound 4t was found to be the most active tyrosinase inhibitor with the IC50 value of 11.3 µM, uncovering that the introduction of the proper hydroxyl group in the 3-aromatic ring was beneficial for enhancing the inhibitory potency against tyrosinase. Moreover, the inhibition mechanism and inhibition kinetics studies revealed that compound 4t presented such inhibitory effect by acting as the reversible and competitive–uncompetitive mixed-II type inhibitor. Further molecular docking simulation showed that 3-aromatic ring of compound 4t was inserted into the narrow regions of binuclear copper-binding site at the bottom of the enzyme binding pocket, while the xanthone skeleton was positioned at the surface of tyrosinase. Taken together, these data suggested that such type of molecules might be utilized for the development of new and promising candidate for the treatment of depigmentation-related disorders.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (2028K) Full view HTML -
Takashi Tsujimoto, Masakazu Nishihara, Yuko Osumi, Takashi Hakamatsuka ...2019Volume 67Issue 11 Pages 1242-1247
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialPolygalaxanthone III, a xanthone glycoside that is a major constituent of “Polygala Root” (Polygala tenuifolia roots, Onji in the Japanese Pharmacopoeia), has been used as a standard in the quality control of crude drugs. However, we previously noted differences in the chromatographic properties of one of three samples of polygalaxanthone III. Therefore, standardization of the standard itself is extremely important. The structures of three standard samples commercially available as polygalaxanthone III were characterized by LC/MS and NMR. LC/MS analysis revealed that two molecular types exist. Both types are chromatographically separable but have an identical mass number with distinguishable MS/MS spectra. One dimensional (1D)-NMR analyses demonstrated that both had the same xanthone moiety and heteronuclear multiple bond correlation (HMBC) analyses revealed that they are structural isomers at the connecting position of glucose to apiose 1-position. Consequently, the isomers were identified as polygalaxanthone III and its regioisomer, polygalaxanthone XI. Based on the findings, we recommend using the LC-MS/MS detection method, which discriminates polygalaxanthone III and XI, to confirm the quality of the standard.
Graphical Abstract Fullsize ImageView full abstractEditor's pickTo guarantee the quality of crude drugs, the marker constituents for quality control corresponding to each crude drug are defined. A standard product with a fixed structure is indispensable for quality control of crude drugs, but the structure of the standard product is unclear in a few cases. The authors found multiple standards for polygalaxanthone III, which is a component of Onji's marker constituent for quality control, and concluded from NMR and MS/MS spectral analyses that their structures were polygalaxanthone III and polygalaxanthone XI. In this study, they propose the use of LC-MS/MS to distinguish polygalaxanthone III and XI.
Download PDF (763K) Full view HTML
-
Hidetoshi Noda, Masakatsu Shibasaki2019Volume 67Issue 11 Pages 1248-1249
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialThe nitrogen inversion energies of a series of N-substituted isoxazolidin-5-ones were studied by density functional theory. The transition state energy was found to strongly correlate with the s-character of the lone pair of electrons on the nitrogen in the ground state. Although the activation energy trends for isxazolidin-5-ones and isoxazolidines are similar, their conformational equilibria are slightly different and the isoxazolidin-5-one inversion energies are generally higher.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (289K) Full view HTML -
Toshinori Suzuki, Rie Takeuchi2019Volume 67Issue 11 Pages 1250-1254
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLMethotrexate is a folate antagonist cytotoxic drug employed in the therapy of cancers and rheumatoid arthritis. Hypobromous acid (HOBr) and hypochlorous acid (HOCl) are generated by eosinophils and neutrophils at inflammation sites. The administered methotrexate may encounter HOBr and HOCl, and react with them to generate products. When methotrexate was incubated with HOBr or HOCl at pH 7.4 and 37°C for 30 min, a single product was generated almost exclusively in each case, identified as 3′-bromomethotrexate for HOBr and 3′-chloromethotrexate for HOCl. When methotrexate was incubated with HOCl in the presence of NaBr, the concentration of 3′-bromomethotrexate increased with decreasing concentration of 3′-chloromethotrexate in a dose-dependent manner with NaBr, probably due to the formation of HOBr. Free amino acids suppressed the reactions of methotrexate with HOBr and HOCl. Taurine suppressed the HOCl reaction but not the HOBr reaction. These results suggest that 3′-bromomethotrexate and 3′-chloromethotrexate may be generated from methotrexate at inflammation sites in humans, although their formation will be suppressed by coexistent amino acids.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (488K) Full view HTML -
Yunong Tian, Lu Feng, Bailin Li, Juanjuan Hu, Jindan Xie, Wenjing Xiao ...2019Volume 67Issue 11 Pages 1255-1258
Published: November 01, 2019
Released on J-STAGE: November 01, 2019
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialOne new 3,24-dinor-2,4-seco-ursane triterpene, rosanortriterpene C (1), together with four known compounds including 24-norursane-type nortriterpenes (2–3), 24-noroleanane-type nortriterpene (4), ursane-type triterpene (5), was isolated from the fruits of Rosa laevigata var. leiocapus. The new structure was elucidated through comprehensive spectroscopic analysis, including one dimensional (1D) and 2D NMR data, as well as electrospray ionization high resolution (HR-ESI) MS and IR spectrometry. Compounds 1–5 showed moderate anti-inflammatory activities against the production of nitric oxide (NO) in RAW264.7 cells stimulated by lipopolysaccharide (LPS) with IC50 values of 10.35 ± 0.92, 14.28 ± 1.20, 5.04 ± 1.43, 29.29 ± 3.64, and 14.37 ± 0.59 µM, respectively.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (392K) Full view HTML
- |<
- <
- 1
- >
- >|