Experimental
ChemistryGeneral Procedure A for the Synthesis of Oleanolic Acid Derivatives (1–15)To OA-OBt (0.2 mmol) and Na2CO3 (0.6 mmol) stirring in DMF (20 mL) was added the corresponding amine (0.2 mmol) or alcohol (0.4 mmol) or methyl ester (0.6 mmol). The mixture was stirred at room temperature for 12 h. After completion (TLC) the solvent was removed under reduced pressure. The mixture was dissolved in EtOAc and washed with water and brine twice. The organic layer was dried over Na2SO4, then filtered and concentrated. The crude product was purified by column chromatography.
General Procedure B for the Synthesis of Oleanolic Acid Derivatives (16–20)To compounds 11–15 (0.1 mmol) stirring in methanol (10 mL) was added 1N NaOH (3 mL). The mixture was stirred at rt. After completion (TLC) the reaction mixture was neutralized with 1 mol/L HCl (3 mL). Water was added and the resulting suspension was filtered. Crude product was purified by column chromatography.
OA-OBtThis compound was prepared from OA (10 mmol), EDCI (13 mmol), (CH3CH2)3N (13 mmol) and 1-hydroxybenzotriazole (HOBt) according to the Lei’s studies.20)
Compound 1This compound was prepared from OA-OBt (0.2 mmol) and ethylenediamine (2 mmol) according to the general procedure A. The residue was purified by column chromatography (dichloromethane/methanol, 15 : 1 v/v). Yield: 62 mg, 62%; white solid. Mp 202.5–203.3°C. 1H-NMR (400 MHz, CDCl3) δ: 0.76, 0.78, 0.92, 0.94, 0.98, 1.18 (7 × CH3), 0.76–2.00 (m, other aliphatic ring protons), 2.66 (d, J = 11.4 Hz, 1H), 2.81 (t, J = 6 Hz, 2H), 3.11–3.21 (m, 2H), 3.36–3.43 (m, 2H), 5.40 (s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 15.00, 15.31, 16.67, 18.07, 23.15, 23.18, 23.27, 25.57, 26.48, 27.07, 27.73, 30.42, 32.26, 32.66 (2C), 33.85, 36.74, 38.35, 38.51, 39.18, 40.44, 40.80, 41.47, 41.69, 46.23, 46.31, 47.35, 55.01, 78.43, 122.79, 144.00, 179.73. Electrospray ionization (ESI)-HRMS (m/z) [M + H]+ Calcd for C32H55N2O2, 499.4258. Found, 499.4257.
Compound 2This compound was prepared from OA-OBt (0.2 mmol) and 1,3-propanediamine (2 mmol) according to the general procedure A. The residue was purified by column chromatography (dichloromethane/methanol, 15 : 1 v/v). Yield: 79 mg, 77%; white solid. Mp 187.6–188.4°C. 1H-NMR (400 MHz, CD3OH) δ: 0.76, 0.78, 0.91, 0.98, 1.17 (7 × CH3), 0.76–2.00 (m, other aliphatic ring protons), 2.56 (d, J = 12.4 Hz, 1H), 2.69 (t, J = 6.56 Hz, 2H), 3.04–3.10 (m, 1H), 3.17–3.21 (m, 1H), 3.37–3.44 (m, 1H), 5.33 (s, 1H), 5.38 (s, 1H), 7.38 (s, 1H); 13C-NMR (100 MHz, CD3OH) δ: 15.90, 16.32, 17.93, 19.48, 23.98, 24.03, 24.56, 26.49, 27.86, 28.54, 28.76, 31.62, 33.10, 33.56, 33.88, 34.50, 35.12, 37.91, 38.15, 39.81, 39.84, 40.68, 42.55, 42.93, 47.55, 47.67, 56.71, 79.66, 124.03, 145.27, 180.48. ESI-HRMS (m/z) [M + H]+ Calcd for C33H57N2O2, 513.4415. Found, 513.4413.
Compound 3This compound was prepared from OA-OBt (0.2 mmol) and 1,4-diaminobutane (2 mmol) according to the general procedure A. The residue was purified by column chromatography (dichloromethane/methanol, 15 : 1 v/v). Yield: 80 mg, 76%; white solid. Mp 179.3–180.2°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.66, 0.84, 0.86, 0.87, 0.88, 1.07 (7 × CH3), 0.66–2.00 (m, other aliphatic ring protons), 2.77 (d, J = 13.8 Hz, 1H), 2.89–3.05 (m, 6H), 5.20 (s, 1H), 7.24 (t, J = 5.4 Hz, 1H), 8.31 (s, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.08, 16.00, 16.81, 18.01, 22.27, 22.92, 23.56, 25.66, 26.59, 26.95(2C), 28.22, 30.41, 30.54, 32.45, 32.74, 32.95, 33.66, 36.57, 38.08, 38.36, 38.74, 40.45, 41.23, 41.27, 45.15, 46.04, 47.12, 54.83, 76.80, 79.18, 121.34, 144.12, 176.01. ESI-HRMS (m/z) [M + H]+ Calcd for C34H59N2O2, 527.4571. Found, 527.4572.
Compound 4This compound was prepared from OA-OBt (0.2 mmol) and 1,5-pentanediamine (2 mmol) according to the general procedure A. The residue was purified by column chromatography (dichloromethane/methanol, 15 : 1 v/v). Yield: 94 mg, 86%; white solid. Mp 142.3–143.2°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.65, 0.66, 0.83, 0.85, 0.87, 0.88, 1.07 (7 × CH3), 0.65–2.00 (m, other aliphatic ring protons), 2.54–2.56 (m, 2H), 2.77 (d, J = 9.54 Hz, 1H), 2.87–3.05 (m, 4H), 5.20 (t, J = 4.92 Hz, 1H), 7.24 (t, J = 5.64 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.15, 16.10, 16.90, 18.06, 22.30, 22.99, 23.64, 23.92, 25.72, 27.00, 28.30, 28.96, 30.49, 31.21, 32.53, 32.82, 33.02, 33.72, 36.63, 38.13, 38.44, 38.84, 38.96, 40.06, 40.50, 40.80, 41.30, 45.25, 46.11, 47.17, 54.88, 76.89, 121.40, 144.20, 176.18. ESI-HRMS (m/z) [M + H]+ Calcd for C35H61N2O2, 541.4728. Found, 541.4728.
Compound 5This compound was prepared from OA-OBt (0.2 mmol) and 1,6-hexylenediamime (2 mmol) according to the general procedure A. The residue was purified by column chromatography (dichloromethane/methanol, 15 : 1 v/v). Yield: 86 mg, 77%; white solid. Mp 138.9–139.5°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.67, 0.85, 0.87, 0.88, 0.89, 1.09 (7 × CH3) 0.67–2.00 (m, other aliphatic ring protons), 2.6 (t, J = 6.8 Hz, 2H), 2.79 (d, J = 10.64 Hz, 1H), 2.91–3.07 (m, 4H), 5.21 (br s, 1H), 5.76 (s, 1H), 7.22 (t, J = 5.52 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.07, 16.03, 16.85, 18.00, 22.27, 22.93, 23.58, 25.66, 26.02, 26.43, 26.95, 28.23, 29.09, 30.84, 32.47, 32.77, 32.95, 33.66, 38.07, 38.76, 39.78, 40.43(2C), 40.46, 46.06, 47.11, 54.81, 76.81, 121.32. ESI-HRMS (m/z) [M + H]+ Calcd for C36H63N2O2, 555.4884. Found, 555.4883.
Compound 6This compound was prepared from OA-OBt (0.2 mmol) and 2-aminoethanol (0.4 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 1 : 1 v/v). Yield: 76 mg, 76%; white solid. Mp 232.4–233.8°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.67, 0.84, 0.86, 0.87, 0.89, 1.08 (7 × CH3), 0.67–2.00 (m, other aliphatic ring protons), 2.75 (d, J = 9.28 Hz, 1H), 2.943.02 (m, 2H), 3.12–3.40 (m, 1H), 3.33–3.36 (m, 2H), 4.27 (d, J = 5.16 Hz, 1H), 4.59 (t, J = 5.32 Hz, 1H), 5.21 (br s, 1H), 7.15 (t, J = 5.56 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.08, 16.01, 16.72, 17.97, 22.34, 22.91, 23.51, 25.64, 26.93(3C), 28.21, 30.41, 32.36, 32.68, 32.89, 33.61, 36.55, 38.36, 40.54, 41.23, 41.54(2C), 45.24, 46.02, 47.09, 54.78, 59.84, 76.80, 121.48, 144.00, 176.50. ESI-HRMS (m/z) [M + Na]+ Calcd for C32H53NO3Na, 522.3918. Found, 522.3919.
Compound 7This compound was prepared from OA-OBt (0.2 mmol) and 3-aminopropanol (0.4 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 1 : 1 v/v). Yield: 80 mg, 77%; white solid. Mp 210.9–211.3°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.67, 0.84, 0.86, 0.88, 0.89, 1.08 (7 × CH3), 0.67–2.00 (m, other aliphatic ring protons), 2.76 (d, J = 9.84 Hz, 1H), 2.96–3.04 (m, 2H), 3.07–3.16 (m, 1H), 3.38 (q, J = 5.44 Hz, 2H), 4.27 (d, J = 5.12 Hz, 1H), 4.41 (t, J = 5.24 Hz, 1H), 5.21 (br s, 1H), 7.22 (t, J = 5.56 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.06, 15.99, 16.79, 17.97, 22.27, 22.90, 23.51, 25.63, 26.93, 28.20, 30.40, 32.25, 32.40, 32.77, 32.89, 33.63, 36.32, 36.55, 38.05, 38.35, 40.06, 40.46(2C), 41.21, 45.17, 46.04, 47.08, 54.78, 58.84, 76.80, 121.43, 144.01, 176.21. ESI-HRMS (m/z) [M + Na]+ Calcd for C33H55NO3Na, 536.4074. Found, 536.4074.
Compound 8This compound was prepared from OA-OBt (0.2 mmol) and 4-amino-1-butanol (0.4 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 1 : 1 v/v). Yield: 86 mg, 81%; white solid. Mp 205.7–206.9°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.66, 0.67, 0.84, 0.86, 0.88, 0.89, 1.08 (7 × CH3), 0.66–2.00 (m, other aliphatic ring protons), 2.78 (d, J = 9.68 Hz, 1H), 2.90–3.08 (m, 3H), 3.38 (q, J = 6 Hz, 2H), 4.28 (d, J = 5.16 Hz, 1H), 4.37 (t, J = 4.96 Hz, 1H), 5.20 (br s, 1H), 7.21 (t, J = 4.96 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.08, 16.00, 16.80, 18.00, 22.25, 22.91, 23.56, 25.66, 25.78, 26.95, 28.22, 30.07, 30.42, 32.45, 32.75, 32.94, 33.66, 36.57, 38.07, 38.36, 38.62, 38.74, 40.44, 41.23, 45.14, 45.16, 46.03, 47.11, 54.81, 60.54, 76.82, 121.34, 144.11, 176.00. ESI-HRMS (m/z) [M + Na]+ Calcd for C34H57NO3Na, 550.4231. Found, 550.4231.
Compound 9This compound was prepared from OA-OBt (0.2 mmol) and 5-amino-1-pentanol (0.4 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 1 : 1 v/v). Yield: 74 mg, 68%; white solid. Mp 195.9–200.9°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.69, 0.84, 0.86, 0.88, 0.89, 1.08 (7 × CH3), 0.67–2.00 (m, other aliphatic ring protons), 2.78 (d, J = 10.2 Hz, 1H), 2.93–3.03 (m, 3H), 3.17 (d, J = 5.36 Hz, 1H), 3.31–3.38 (m, 3H), 4.28 (d, J = 3.96 Hz, 1H), 4.33 (t, J = 5.04 Hz, 1H), 5.20 (s, 1H), 7.19 (t, J = 5.24 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.07, 16.01, 16.82, 18.01, 22.26, 22.92, 23.10, 23.56, 25.66, 26.95, 28.23, 29.01, 30.42, 32.25, 32.30, 32.47, 32.75, 32.95, 33.67, 36.57, 38.08, 38.37, 40.46, 41.24, 45.16, 46.05, 47.12, 54.83, 60.58, 60.70, 76.70, 76.82, 121.34, 144.12, 176.00. ESI-HRMS (m/z) [M + Na]+ Calcd for C35H59NO3Na, 564.4387. Found, 564.4385.
Compound 10This compound was prepared from OA-OBt (0.2 mmol) and 6-amino-1-hexanol (0.4 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 1 : 1 v/v). Yield: 90 mg, 80%; white solid. Mp 183.8–184.5°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.66, 0.67, 0.84, 0.86, 0.88, 0.89, 1.08 (7 × CH3), 0.66–2.00 (m, other aliphatic ring protons), 2.78 (dd, J = 3.96, 13.38 Hz, 1H), 2.92–3.04 (m, 3H), 3.36 (dd, J = 6.6, 11.76 Hz, 2H), 4.29 (d, J = 5.22 Hz, 1H), 4.33 (t, J = 5.16 Hz, 1H), 5.20 (br s, 1H), 7.20 (t, J = 5.52 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.07, 16.04, 16.85, 18.00, 22.26, 22.94, 23.59, 25.38, 25.67, 26.62, 26.96, 28.24, 29.20, 30.45, 32.47, 32.63, 32.77, 32.97, 33.67, 36.58, 38.08, 38.39, 38.83, 38.91, 40.06, 40.46, 41.25, 45.19, 46.06, 47.12, 54.83, 60.69, 76.83, 121.33, 144.18, 176.02. ESI-HRMS (m/z) [M + Na]+ Calcd for C36H61NO3Na, 578.4544. Found, 578.4545.
Compound 11This compound was prepared from OA-OBt (0.2 mmol) and glycine methyl ester hydrochloride (0.8 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 3 : 1 v/v). Yield: 94 mg, 89%; white solid. Mp 137.8–138.6°C. 1H-NMR (600 MHz, (CD3)2SO) δ: 0.64, 0.67, 0.84, 0.87, 0.88, 0.89, 1.08 (7 × CH3), 0.64–2.00 (m, other aliphatic ring protons), 2.76(d, J = 9.6 Hz, 1H), 2.97–3.00 (m, 1H), 3.59 (s, 3H), 3.64 (dd, J = 5.52 Hz, 17.04 Hz, 1H), 3.80 (dd, J = 6 Hz, 16.98 Hz, 1H), 4.29 (d, J = 5.16 Hz, 1H), 5.17 (t, J = 3.36 Hz, 1H), 7.76 (t, J = 5.7 Hz, 1H); 13C-NMR (150 MHz, (CD3)2SO) δ: 15.13, 16.05, 16.60, 18.02, 22.40, 22.94, 23.50, 25.64, 26.84, 26.97, 28.24, 30.42, 32.42, 32.50, 32.90, 33.61, 36.59, 38.09, 38.40, 38.86, 40.35, 40.93, 41.24, 45.19, 46.05, 47.14, 51.51, 54.83, 76.83, 121.42, 143.95, 170.54, 176.94. ESI-HRMS (m/z) [M + Na]+ Calcd for C33H53NO4Na, 550.3867. Found, 550.3862.
Compound 12This compound was prepared from OA-OBt (0.2 mmol) and methyl 3-aminopropionate hydrochloride (0.8 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 3 : 1 v/v). Yield: 90 mg, 83%; white solid. Mp 218.8–219.6°C. 1H-NMR (600 MHz, (CD3)2SO) δ: 0.65, 0.67, 0.84, 0.86, 0.87, 0.89, 1.07 (7 × CH3), 0.65–2.00 (m, other aliphatic ring protons), 2.40–2.43 (m, 2H), 2.74 (dd, J = 3.96, 13.38 Hz, 1H), 2.97–3.00 (m, 1H), 3.15–3.21 (m, 1H), 3.25–3.21 (m, 1H), 3.57 (s, 3H), 4.28 (d, J = 4.74 Hz, 1H), 5.19 (t, J = 3.42 Hz, 1H), 7.35 (t, J = 5.58 Hz, 1H); 13C-NMR (150 MHz, (CD3)2SO) δ: 15.10, 16.03, 16.77, 17.99, 22.27, 22.92, 23.52, 25.65, 26.86, 26.96, 28.23, 30.42, 32.40, 32.61, 32.92, 33.57, 33.61, 35.02, 36.57, 38.08, 38.38, 38.89, 40.43, 41.22, 45.22, 46.01, 47.10, 51.26, 54.81, 76.82, 121.48, 143.93, 171.93, 176.47. ESI-HRMS (m/z) [M + Na]+ Calcd for C34H55NO4Na, 564.4023. Found, 564.4025.
Compound 13This compound was prepared from OA-OBt (0.2 mmol) and methyl 4-aminobutyrate hydrochloride (0.8 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 3 : 1 v/v). Yield: 87 mg, 78%; white solid. Mp 190.4–191.7°C. 1H-NMR (400 MHz (CD3)2SO) δ: 0.54, 0.64, 0.67, 0.84, 0.86, 0.89, 1.08 (7 × CH3), 0.54–1.97 (m, other aliphatic ring protons), 2.27 (t, J = 7.48 Hz, 2H), 2.78 (d, J = 10.00 Hz, 1H), 2.97–3.04 (m, 3H), 3.58 (s, 3H), 4.28 (d, J = 5.08 Hz, 1H), 5.21 (s, 1H), 7.31 (t, J = 5.72 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.12, 16.08, 16.86, 18.08, 22.27, 22.98, 23.66 (2C), 24.47, 25.77, 27.03 (2C), 28.30, 30.51, 30.93, 32.53, 32.87, 33.03, 33.71 (2C), 36.64, 38.16, 38.20, 38.45, 40.52, 41.30, 45.32, 46.07, 47.19, 51.28, 54.90, 76.90, 121.47, 144.16, 173.22, 176.33. ESI-HRMS (m/z) [M + Na]+ Calcd for C35H57NO4Na, 578.4180. Found, 578.4183.
Compound 14This compound was prepared from OA-OBt (0.2 mmol) and methyl 5-aminopentanoate hydrochloride (0.8 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 3 : 1 v/v). Yield: 77 mg, 67%; white solid. Mp 178.8–179.6°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.65, 0.67, 0.84, 0.86, 0.88, 0.89, 1.08 (7 × CH3), 0.65–2.00 (m, other aliphatic ring protons), 2.27 (t, J = 7.24 Hz, 2H), 2.76 (d, J = 9.84 Hz, 1H), 2.93–3.05 (m, 3H), 3.57 (s, 3H), 4.26 (d, J = 5.16 Hz, 1H), 5.20 (s, 1H), 7.24 (t, J = 5.52 Hz, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.01, 15.99, 16.79, 17.97, 22.01, 22.22, 22.87, 23.55, 25.64, 26.94, 28.21, 28.50, 30.42, 32.43, 32.77, 32.93, 33.02(2C), 33.63, 36.55, 38.06, 38.31, 38.35, 38.88, 40.44, 41.22, 45.19, 46.02, 47.10, 51.11, 54.79, 76.80, 121.36, 144.08, 173.21, 176.08. ESI-HRMS (m/z) [M + Na]+ Calcd for C36H59NO4Na, 592.4336. Found, 592.4333.
Compound 15This compound was prepared from OA-OBt (0.2 mmol) and methyl 6-aminocappoate hydrochloride (0.8 mmol) according to the general procedure A. The residue was purified by column chromatography (petroleum ether/EtOAc, 3 : 1 v/v). Yield: 95 mg, 81%; white solid. Mp 116.2–117.4°C. 1H-NMR (600 MHz (CD3)2SO) δ: 0.66, 0.67, 0.84, 0.86, 0.87, 0.89, 1.08 (7 × CH3), 0.66–2.00 (m, other aliphatic ring protons), 2.27 (t, J = 7.44 Hz, 2H), 2.77 (dd, J = 4.02, 13.44 Hz, 1H), 2.91–3.04 (m, 3H), 3.57 (s, 3H), 4.27 (d, J = 5.16 Hz, 1H), 5.20 (t, J = 3.42 Hz, 2H), 7.21 (t, J = 5.58 Hz, 2H). 13C-NMR (150 MHz, (CD3)2SO) δ: 15.06, 16.03, 16.85, 18.00, 22.26, 22.93, 23.59, 24.23, 25.67, 26.06, 26.96, 28.24, 28.77, 30.45, 32.46, 32.77, 32.97, 33.30, 33.66, 36.58, 38.08, 38.39, 38.60, 38.91, 40.06, 40.46, 41.25, 45.20, 46.05, 47.12, 51.17, 54.82, 76.83, 121.34, 144.17, 173.26, 176.07. ESI-HRMS (m/z) [M + Na]+ Calcd for C37H61NO4Na, 606.4493. Found, 606.4493.
Compound 16This compound was prepared from 11 (0.2 mmol) and sodium hydroxide (0.6 mmol) according to the general procedure B. Yield: 84 mg, 82%; white solid. Mp 176.0–177.8°C. 1H-NMR (600 MHz, (CD3)2SO) δ: 0.64, 0.67, 0.83, 0.87, 0.87, 0.89, 1.08 (7 × CH3), 0.64–2.00 (m, other aliphatic ring protons), 2.76 (dd, J = 3.84, 13.38 Hz, 1H), 2.97–3.00 (m, 1H), 3.56 (dd, J = 3.54, 17.28 Hz, 1H), 3.73 (dd, J = 6.0, 17.28 Hz, 1H), 4.28 (d, J = 4.44 Hz, 1H), 5.19 (t, J = 3.36 Hz, 1H), 7.57 (t, J = 5.58 Hz, 1H), 12.37 (s, 1H); 13C-NMR (150 MHz, (CD3)2SO) δ: 15.12, 16.05, 16.60, 18.00, 22.45, 22.95, 23.50, 25.65, 26.85, 26.97, 28.24, 30.42, 32.39, 32.48, 32.91, 33.62, 36.58, 38.10, 38.39, 38.88, 40.47, 40.90, 41.24, 45.14, 46.05, 47.15, 54.82, 76.83, 121.48, 143.97, 171.48, 176.68. ESI-HRMS (m/z) [M + Na]+ Calcd for C32H51NO4Na, 536.3710. Found, 536.3713.
Compound 17This compound was prepared from 12 (0.2 mmol) and sodium hydroxide (0.6 mmol) according to the general procedure B. Yield: 90 mg, 85%; white solid. Mp >250°C. 1H-NMR (600 MHz (CD3)2SO) δ: 0.65, 0.67, 0.84, 0.86, 0.86, 0.88, 1.07 (7 × CH3), 0.65–2.00 (m, other aliphatic ring protons), 2.33 (t, J = 7.08 Hz, 2H), 2.72 (dd, J = 3.78, 13.80 Hz, 1H), 2.97–3.00 (m, 1H), 3.123.15 (m, 1H), 3.23–3.29 (m, 1H), 3.37 (s, 1H), 5.19 (br s, 1H), 7.30 (t, J = 5.58 Hz, 1H), 12.20 (s,1H); 13C-NMR (150 MHz, (CD3)2SO) δ: 15.16, 16.09, 16.83, 18.05, 22.33, 22.97, 23.55, 25.69, 26.93, 27.00, 28.28, 30.47, 32.43, 32.66, 32.96, 33.64, 33.82, 35.02, 36.61, 38.13, 38.43, 38.95, 40.56, 41.28, 45.23, 46.05, 47.15, 54.84, 76.87, 121.62, 143.93, 173.21, 176.44. ESI-HRMS (m/z) [M + Na]+ Calcd for C33H53NO4Na, 550.3867. Found, 550.3865.
Compound 18This compound was prepared from 13 (0.2 mmol) and sodium hydroxide (0.6 mmol) according to the general procedure B. Yield: 94 mg, 87%; white solid. Mp 228.7–229.5°C. 1H-NMR (400 MHz, (CD3)2SO) δ: 0.65, 0.67, 0.84, 0.86, 0.89, 1.08 (7 × CH3), 0.65–2.00 (m, other aliphatic ring protons), 2.18 (t, J = 7.4 Hz, 2H), 2.78 (d, J = 9.68 Hz, 1H), 2.96–3.03 (m, 3H), 3.32 (d, J = 8.92 Hz, 2H), 4.28 (d, J = 5.08 Hz, 1H), 5.21 (brs, 1H), 7.29 (t, J = 5.44 Hz, 1H), 12.0 (s, 1H); 13C-NMR (100 MHz, (CD3)2SO) δ: 15.16, 16.10, 16.92, 18.08, 22.27, 22.99, 23.67, 24.53, 25.76, 27.04, 28.31, 30.52, 31.29, 32.53, 32.87, 33.03, 33.71, 36.65, 38.23, 38.43, 38.46, 40.50, 41.31, 45.29, 45.30, 46.09, 47.19, 54.89, 76.79, 76.90, 121.48, 144.17, 174.36, 176.24. ESI-HRMS (m/z) [M + Na]+ Calcd for C34H55NO4Na, 564.4023. Found, 564.4024.
Compound 19This compound was prepared from 14 (0.2 mmol) and sodium hydroxide (0.6 mmol) according to the general procedure B. Yield: 79 mg, 71%; white solid. Mp 139.1–140.9°C. 1H-NMR (600 MHz (CD3)2SO) δ: 0.65, 0.66, 0.83, 0.86, 0.87, 0.88, 1.07 (7 × CH3), 0.65–2.00 (m, other aliphatic ring protons), 2.17 (t, J = 7.26 Hz, 2H), 2.78 (dd, J = 4.08, 13.56 Hz, 1H), 2.94–3.02 (m, 3H), 3.41 (s, 1H), 5.20 (br s, 1H), 7.26 (t, J = 5.52 Hz, 1H), 11.98 (s, 1H); 13C-NMR (150 MHz, (CD3)2SO) δ: 15.16, 16.09, 16.83, 18.05, 22.33, 22.97, 23.55, 25.69, 26.93, 27.00, 28.28, 30.47, 32.43, 32.66, 32.96 (2C), 33.64, 33.82, 35.02, 36.61, 38.13, 38.43, 38.95, 40.06, 40.56, 41.28, 45.23, 46.05, 47.15, 54.84, 76.87, 121.62, 143.93, 173.21, 176.44. ESI-HRMS (m/z) [M + Na]+ Calcd for C35H57NO4Na, 578.4180. Found, 578.4180.
Compound 20This compound was prepared from 15 (0.2 mmol) and sodium hydroxide (0.6 mmol) according to the general procedure B. Yield: 82 mg, 72%; white solid. Mp 119.8–120.7°C. 1H-NMR (600 MHz, (CD3)2SO) δ: 0.66, 0.67, 0.84, 0.86, 0.87, 0.88, 1.08 (7 × CH3), 0.66–2.00 (m, other aliphatic ring protons), 2.17 (t, J = 7.38 Hz, 2H), 2.78 (dd, J = 3.9, 13.32 Hz, 1H), 2.91–3.04 (m, 3H), 5.20 (brs, 1H), 7.22 (t, J = 5.58 Hz, 2H), 11.98 (s, 1H); 13C-NMR (150 MHz, (CD3)2SO) δ: 15.08, 16.05, 16.86, 18.01, 22.27, 22.94, 23.60, 24.30, 25.68, 26.17, 26.97, 28.25, 28.86, 30.46, 32.47, 32.77, 32.98, 33.67(2C), 36.59, 38.08, 38.39, 38.66, 38.91, 40.05, 40.47, 41.26, 45.20, 46.06, 47.13, 54.83, 76.84, 121.35, 144.16, 174.40, 176.06. ESI-HRMS (m/z) [M + Na]+ Calcd for C36H59NO4Na, 592.4346. Found, 592.4344.
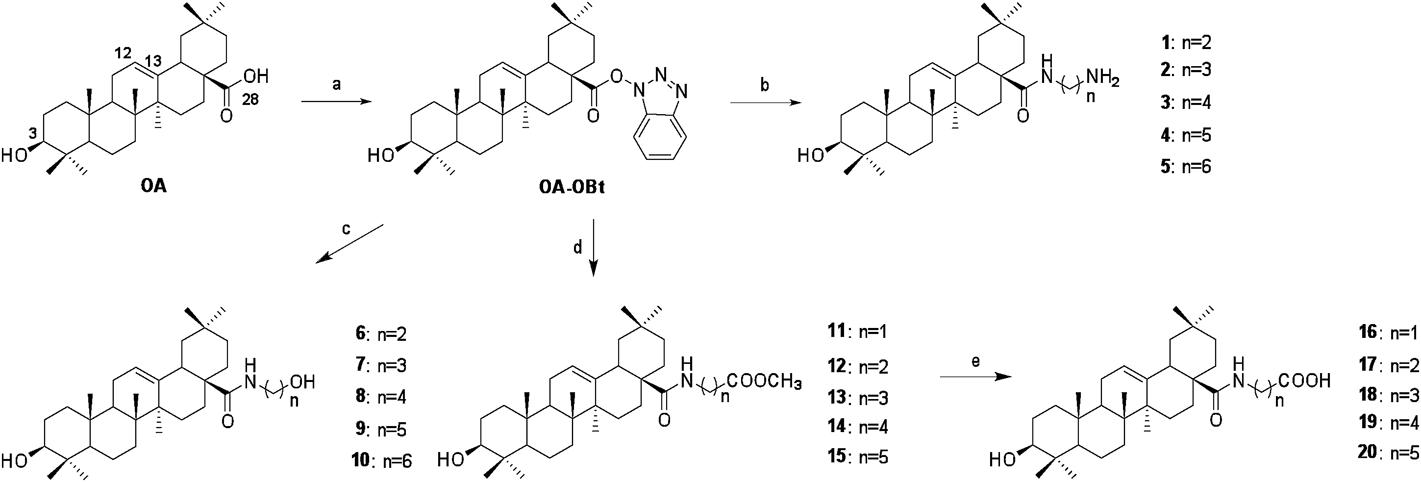
BioassaysCell Activity AssayMDCK cells were seeded into 96-well plates in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) cultured overnight at 37°C in 5% CO2. Then, cells were suspended in DMEM supplemented with 1% FBS, containing test compound and 2 µg/mL TPCK-treated trypsin, and the cells were further incubated at 37°C in 5% CO2 for 40 h. Cell viability was assessed using the CellTiter-Glo assay kit (Promega Corp., Madison, WI, U.S.A.) as recommended by the supplier, and the plates were read using a plate reader (Molecular Devices SpectraMax M2). Viability was calculated using the background corrected absorbance as follows: Viability (%) = (CA of experiment well/DA of control well) × 100, where CA, DA represent the reading values of test compound and DMSO, respectively.
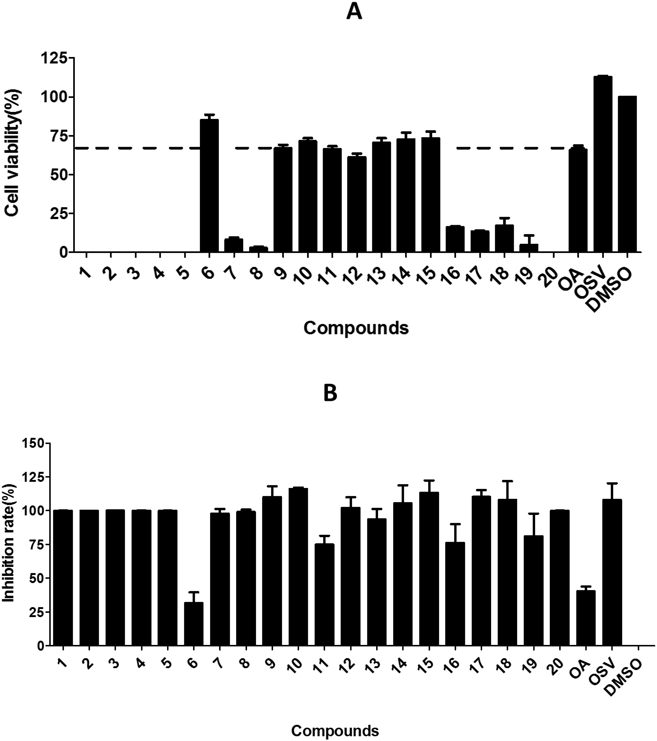
Cytopathic Effect (CPE) Reduction AssayMDCK cells were seeded into 96-well plates, incubated overnight and infected with influenza virus (MOI = 0.1) suspended in DMEM supplemented with 1% FBS, containing test compound and 2 µg/mL TPCK-treated trypsin, with a final DMSO concentration of 1% in each well. After 40 h of incubation, CellTiter-Glo reagent was added and the plates were read using a plate reader (Molecular Devices SpectraMax M2).
Hemagglutination Inhibition (HI) AssayCompound from a 2-fold serial dilution in saline was mixed with an equal volume of influenza virus (The HA titers of A/WSN/33 (H1N1) virus is 1 : 2) in the V-bottomed 96-well microplate. Subsequently, 50 µL of freshly prepared chicken RBCs (1% v/v in saline) were added to each well. The mixture was incubated for 30 min at RT before observing RBCs aggregation on the plate.
SPRInteractions between the influenza HA and the compounds were analyzed using the Biacore T200 system (GE Healthcare, Uppsala, Sweden) at 25°C. Recombinant influenza HA (Sino Biological Inc., Beijing, China) was immobilized on a sensor chip (CM5) using an amine coupling kit (GE Healthcare, Buckinghamshire, U.K.). Final HA-immobilized levels were typically approx. 16000 RU. Subsequently, compounds were injected as analytes at various concentrations, and PBS-P (10 mM phosphate buffer with 2.7 mM KCl and 137 mM NaCl, 0.05% surfactant P20, pH 4.5) was used as running buffer. For binding studies, analytes were applied at corresponding concentrations in running buffer at a flow rate of 30 mL/min with a contact time of 60s and a dissociation time of 60s. Chip platforms were washed with running buffer and 50% DMSO. Data were analyzed with the Biacore evaluation software (T200 version 1.0) by curve fitting using a binding model of 1 : 1.