Review
Metal-Free Oxidative Cross-Coupling Reaction of Heteroaromatic and Related Compounds
2019 年 67 巻 12 号 p. 1259-1270
詳細
2019 年 67 巻 12 号 p. 1259-1270
The biary unit having heteroatom as important scaffolds widely exist in a large number of biologically active compounds and functional organic molecules. Since the cross-coupling is a useful synthetic method for constructing biaryl and heterobiaryl structures, the development of novel cross-coupling methods has been studied intensively. The oxidative biaryl coupling reaction of aromatic compounds having heteroatoms is an attractive method since they do not require the prefunctionalization of arenes. This report describes recent advances in hypervalent iodine(III) induced metal-free synthesis of biaryls having heteroatoms.
Aromatic compounds with heteroatoms in and on the rings have received considerable attention as building blocks of functional compounds due to their varied applications in the fields of pharmaceutical science, materials science, natural product development, and many others.1) Therefore, the development of cross-coupling reactions of heteroatom-containing aromatic compounds remains the focus of intensive study in organic chemistry.2–5) It is difficult to develop an ideal method to achieve the synthesis of heteroatom-containing biaryl compounds, the development of which is important in modern organic chemistry.6–11) In general, the C–C bond-forming reaction using transition metal catalysts requires the synthesis of functionalized coupling partners as starting materials, such as ArX, ArSnR3, ArBR2, and ArZn12–15) (Chart 1, Eq. 1). In addition, the synthesis of a functionalized starting material often requires several synthetic processes.
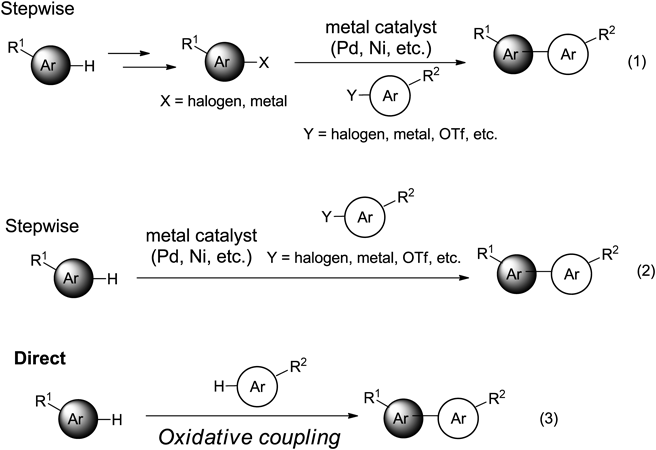
There has been remarkable progress over the past decade in the development of carbon–hydrogen (C–H) cross-coupling with a C–H bond activation method16–20) (Eq. 2). Transition metal-catalyzed C–H activation cross-coupling of aromatic compounds with aryl halides has emerged as an efficient synthetic tool for the construction of functional biaryl compounds.21–27) Recently, novel metal-catalyzed cross-coupling reactions of heteroaromatic compounds have been developed by Gaunt,28) Sames,29,30) Sanford,31) Glorius,32,33) Ackermann,34,35) Itami,36–38) and Larrosa.39,40) However, these reactions have several limitations, such as harsh reaction conditions, the need for metal catalysts, and the use of excessive amounts of substrate.
Theoretically, the most straightforward method for forming C–C bonds is an oxidative synthesis that couples two C–H bonds on a coupling substrate41–45) (Eq. 3). This strategy can be synthesized with less cost or waste compared with previously reported methods, since the process of functionalization of the two coupling substrates is not required. Although it is an oxidative coupling method, which is superior from the viewpoint of green chemistry since the oxidation potential of the product is lower than that of the corresponding starting material, the formation of undesired homo-coupling and over-oxidation occurred. Therefore, the most important issue in this research field is the development of a direct oxidative cross-coupling method for selectively controlling the cross-coupling product. Recently, DeBoef and colleagues46) and Stuart and Fagnou47) have reported their pioneering work on oxidative cross coupling of aromatic compounds. However, there are problems in these reaction methods such as the use of excess substrate, severe reaction conditions (i.e., high temperature), stoichiometric amounts of oxidizing agent, and moderate yields. Therefore, despite this progress, further development of a novel oxidative cross-coupling reaction access to functional biaryl molecules is still highly desirable.
Because of their low toxicity and easy handling, hypervalent iodine reagents have emerged as viable alternatives to toxic heavy-metal reagents.48–54) We have studied novel oxidative bond formation reactions of electron-rich aromatic compounds using hypervalent iodine(III) reagents. As a result, we have developed the direct oxidative nucleophilic substitution of phenyl ethers with various nucleophiles using phenyliodine(III) bis(trifluoroacetate) (PIFA).55–57) In further study, a combination of Lewis acid and PIFA was used to identify a biaryl coupling reaction of phenyl ether and alkyl arene via a cation radical intermediate.58–63) The coupling reaction proceeds via cation radicals with the neutral molecule of the substrate to obtain a coupling product. We have recently reported hypervalent iodine(III) reagent induced oxidative cross-coupling reactions of aromatic compounds64–69) (Chart 2).
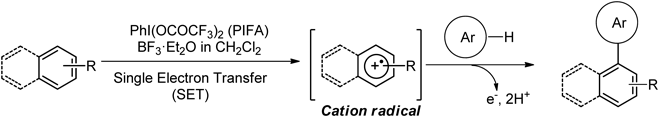
This report describes recent advances in the metal-free synthesis of biaryls having heteroatoms, such as thiophene, pyrrole and phenol, using hypervalent iodine(III) reagents. It also describes recent progress in glycosylation reactions using hypervalent iodine reagents.
Polythiophenes have attracted much attention due to their optical, electrical and electrochemical properties.70–72) Because the oxidation potentials of 2,2′-bithiophenes are lower than thiophenes, they can be polymerized under mild conditions, so that higher quality α-linked polymers can be synthesized from these than those prepared from thiophenes.71) However, it is difficult to control the reaction, and there has been no report of synthesis using oxidative coupling of thiophene due to the low oxidation potential of bithiophenes. Thus, bithiophene usually causes over-oxidation to produce oligomeric polythiophenes.73) We have succeeded in developing coupling reactions using hypervalent iodine(III) reagents to substituted thiophenes74,75) (Chart 3). However, these methods required excess substrate at low temperatures, and the product was a mixture of inseparable regioisomers. In the case of 3-substituted pyrrole, the coupling product could not be synthesized regioselectively.76,77) These reactions proceeded via cation radical intermediates to obtain coupling products. Thus, the regioselective coupling reaction of 3-substituted heteroaromatic compounds was not achieved.
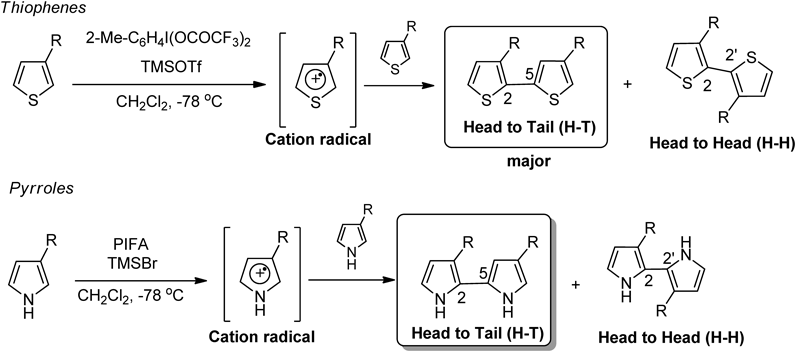
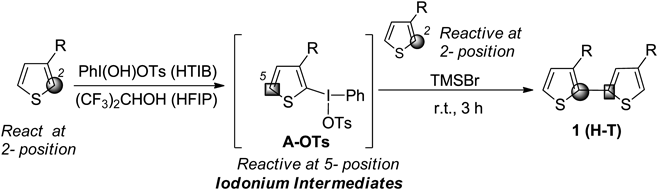
Therefore, the regioselective synthesis of H-T dimers of 3-substituted thiophene was examined. In the case of the coupling of 3-substituted thiophenes, the resulting coupling product produces three regioisomers. Among these, synthesis of the head-tail dimer, which is unsymmetrical between the 2- and 5-position of the thiophene ring, is considered to be the most difficult. In 2007, our group reported the synthesis of the diaryl iodonium(III) salts of a heteroaromatic compound.78,79) Therefore, the reaction using heteroaromatic iodonium(III) salts was investigated to achieve regioselective coupling reactions. After optimization, the coupling reaction of thiophene proceeded through the iodonium intermediate. The formation of other isomers was not observed; H-T dimer as a single isomer was obtained80,81) (Chart 4).
The substrate scope is shown in Chart 5. 3-Alkyl thiophenes gave H-T dimers with high regioselectivity regardless of the length of the alkyl groups. The reaction also proceeded with the steric hindrance substrate, and gave H-T dimers 1e (H-T) and 1f (H-T). The bromo group was tolerable during the reaction, and the product was obtained with high regoioselectivity.
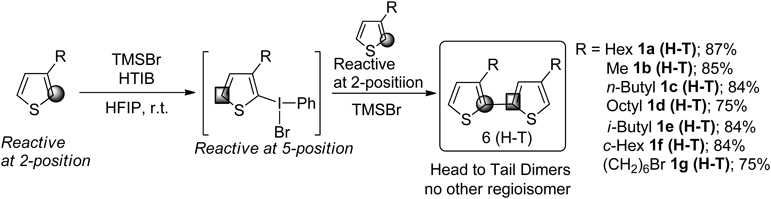
Next, we examined the cross-coupling reactions of various heteroaromatic compounds such as thiophene, pyrrole and indole. Based on our studies for the case of heteroaromatic compounds, the cross-coupling products were obtained in a low yield, and the undesirable homo-coupling reaction proceeded. The heteroaromatic compounds having different reactive sites are too sensitive to react selectively even through the single electron transfer (SET) oxidation processes using the hypervalent iodine reagents. On the other hand, as shown in Chart 6, regioselective synthesis of H-T dimers of thiophenes could be achieved via an iodonium(III) intermediate. Therefore, the oxidative cross-coupling reactions of heteroaromatic compounds using these iodonium salts as reaction intermediates were examined.
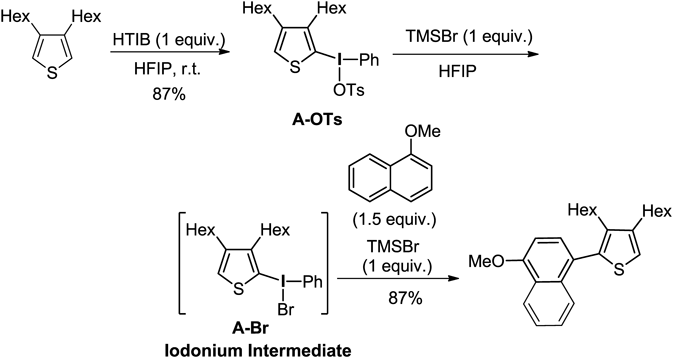
The cross-coupling reaction with 1-methoxynaphthalene was investigated using isolatable α-thienyl iodonium(III) salt A-OTs having a tosylate anion. As a result of the addition of various acids and heating, the reaction did not proceed, and the stable salt A-OTs was recovered from the reaction mixture. However, as a result of further examination of the additive, in the case of bromotrimethylsilane (TMSBr), the reaction of the iodonium salt A-OTs resulted in the formation of a cross-coupling product in a low yield. The solvents were then evaluated using TMSBr. As a result, when hexafluoroisopropanol (HFIP) was used, the yield of the cross-coupling product was improved, and the product was obtained in a yield of 87% (Chart 6). The use of 2 eq of TMSBr to the iodonium salt A-OTs was necessary to perform the reaction. When one equivalent of TMSBr was used, the iodonium bromide A-Br was produced quantitatively. Therefore, in our cross-coupling reaction, TMSBr should be used for the in situ formation of the iodonium bromide A-Br as a reaction intermediate.82,83)
The cross-coupling reaction between heteroaromatic compounds with electron-rich aromatic arenes proceeded without the formation of undesirable homodimers or over-oxidation (Chart 7). As the coupling partners to these salts, the nucleophilic properties of arenes are important factors in this cross-coupling reaction, and various aromatic nucleophiles can be used for the reaction, such as phenyl ethers and heteroaromatic compounds. In the case of thiophene–pyrrole, the reaction proceeded selectively to give the cross-coupling product 2d. Selective cross-coupling between arenes with similar properties, with iodonium salts as intermediates, was performed. The coupling reaction proceeded smoothly without by-production of the homo-coupling, producing good yield. As shown in product 2h, the halogen functionality remains unreacted during the reaction, which allows further modification of the mixed biaryl structure obtained using the reported bond-formation reaction.
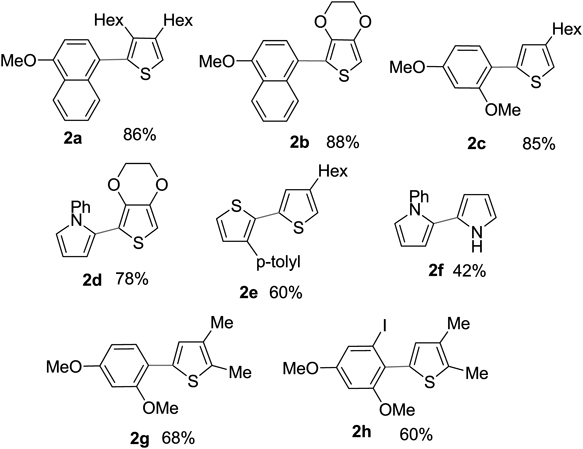
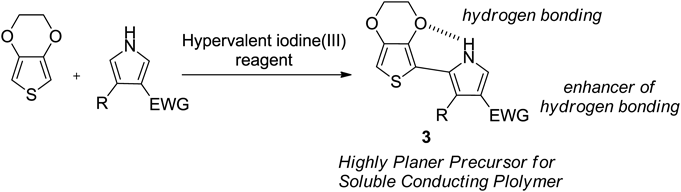
Thienyl-pyrrole and their derivatives are considered useful precursors because they can control their properties as corresponding polymers by introducing substituents on the pyrrole nitrogen atom.84–86) On the other hand, poly(3,4-ethylenedioxythiophene) (PEDOT)87–92) and polypyrrole are widely used as conductive polymers because of their useful properties.93–95) Therefore, we envisaged that hypervalent iodine(III) reagents would enable the oxidative coupling reaction for the synthesis of EDOT-pyrrole 396) (Chart 8).
Based on our research results, we performed the coupling reaction of EDOT with various pyrroles. As shown in Chart 9, this resulted in coupling products 3a–3c being obtained in good yields. Ester substituted pyrroles also gave coupling products 3e to 3g in good yield. Pyrroles substituted with the electron-withdrawing groups cyano and sulfonyl as acceptor substrates gave the coupling products 3h–3j, respectively.
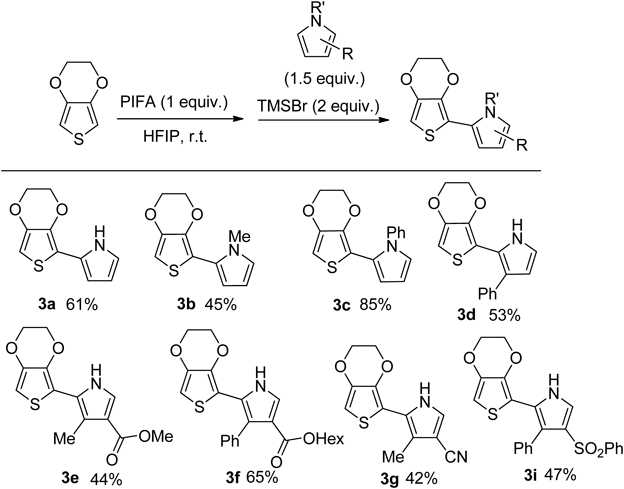
The structure of compound 3e was confirmed by single crystal X-ray analysis. As a result, the distance between N5 of 2.764 Å pyrrole and O3 of EDOT is shorter than the sum (3.07 Å) of the van der Waals radii of nitrogen and oxygen. That is, it was found that the strong hydrogen bond between the EDOT oxygen atom and the pyrrole NH group makes the thiophene ring and the pyrrole ring have a completely planar anticonformation. This O–H–N hydrogen bonding interaction is important for the self-planarization and stiffening of π-conjugated systems.
(Color figure can be accessed in the online version.)
The pyrrole–aryl backbone is an important structural attribute, as well as an important structural motif of fluorescent dyes, organic conductive materials, and biologically active compounds.97–99) Therefore, the development of efficient synthetic methods for such pyrrole–aryls is of significant importance in the field of synthetic chemistry. We assumed that the stabilized pyrrolyl iodonium(III) salt could enable the cross-coupling of pyrrole. We found a direct cross-coupling reaction of pyrroles with aromatic compounds via stabilized pyrrolyl iodonium(III) salt100) (Chart 10).
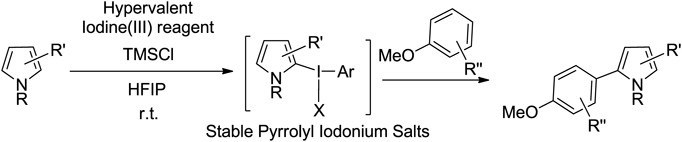
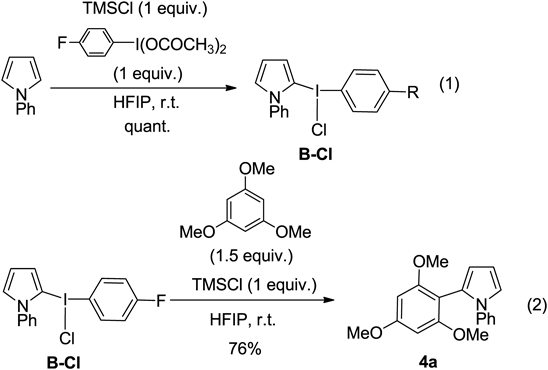
As a result of optimizing the reaction conditions, the best results were obtained when TMSCl was used with 4-furuorophenyliodine diacetate (4-F-PIDA) to form the pyrrole cross-coupling product (Chart 11, Eq. 1). To elucidate the reaction mechanism, it was found that when the combination with 4-F-PIDA and TMSCl was used, the iodonium intermediate B-Cl was produced in quantitative yield. The pyrrolyl iodonium salt B-Cl was then reacted with 1,3,5-trimethoxybenzene and TMSCl to afford the corresponding coupling product 4a in 76% yield (Eq. 2). From these results, it was determined that TMSCl and 4-F-PIDA are the best and most specific reagents for this coupling reaction.
With these optimized conditions, the reaction scope was explored in terms of the cross-coupling reaction of pyrrole in one pot. The cross-coupling product 4 was obtained by the reaction of pyrrole with arenes, the results of which are shown in Chart 12. In this coupling reaction, only the C-2 arylated product 4 was formed. In the case of N-pheny pyrroles, the reaction proceeded smoothly and gave the expected coupling product 4b. Steric limitation had no effect on this reaction. The reaction of pyrroles with various arenes was able to undergo the coupling reaction to give the corresponding coupling products 4c–4f in good to moderate yields. When we performed the reaction of 3-substituted pyrrole, the reaction proceeded smoothly with high regioselectivity, and 4g and 4h of the coupling products were obtained as single isomers. The reaction of the 3-dialkyl-substituted pyrrole also proceeded, and the coupled product 4i was produced in 50% yield.
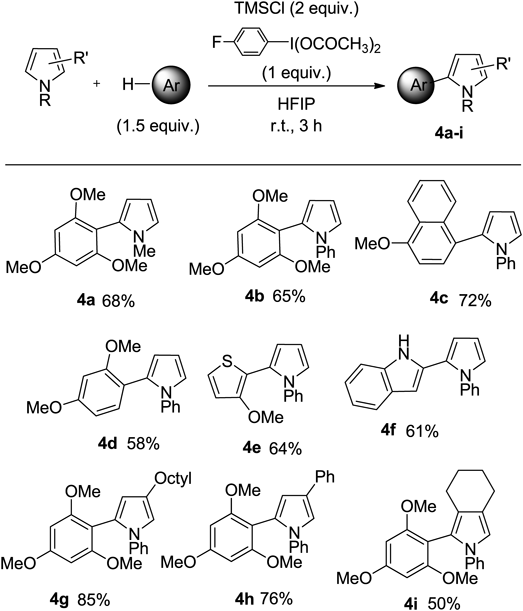
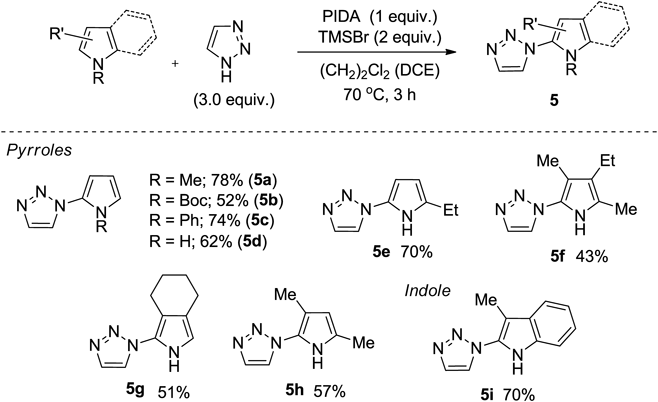
The coupling reaction that proceeds with pyrrolyl iodonium salt as an intermediate can be applied to the synthesis of N-pyrrolylazoles. The N-pyrrolylazoles serve as an important structural core of various natural products and functional materials.101–103) However, due to the instability of pyrrole based metals and halides, conventional approaches to N-aryl azoles have been limited to methods such as Ullmann coupling.104–106) Therefore, we examined the application of our coupling reaction pyrrole with azoles. As a result, when TMSBr was used in combination with PIDA, the best results were obtained in forming N-pyrrolylazole, and the C–N-coupling reaction proceeded in good yield.107,108)
The scope of the substrate for our method proceeded without being affected by the N-substituent, and products 5 were obtained in good yield (Chart 13). N-Free pyrrole and 2-ethyl pyrrole afforded the corresponding coupling products 5d and 5e in considerable yields. In the case of poly-substituted pyrroles, the coupling products 5f, 5g, and 5h were obtained in moderate yields. The reaction proceeded when carried out with 3-methylindole, and the coupling product 5i was obtained in a yield of 70%.
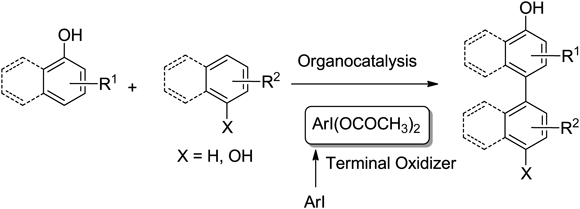
The oxidative cross-coupling of phenols has attracted significant interest from organic chemists due to its environmentally friendly methods and economic advantages.109–111) For the oxidative homo-coupling reactions of phenols, efficient methods have been reported, including the use of Fe, Cu, Mn and Ti salts.112–115) However, the cross-coupling reaction of phenols remains challenging due to the difficulty of controlling undesirable homo-coupling as a byproduct.116–119) In addition, the formation of high molecular-weight polymers or C–O-bond fomation reactions also proceeds readily under oxidative conditions.120,121) This is because the oxidation potential of phenol cannot be adjusted by introducing a protective group into the hydroxyl group, making it difficult to control its reactivity. Therefore, the development of a general method for the cross-coupling reaction of phenols is highly desirable. The cross-coupling reaction of phenols using a boron-doped diamond electrode has been reported by Waldvogel and colleagues122) (Chart 14). Recently, an iron-catalyzed oxidative cross-coupling reaction of phenols has also been developed.123) Although metal-catalyzed cross-coupling of phenols has been studied, the oxidative cross-coupling reaction using organooxidants is still extremely challenging.

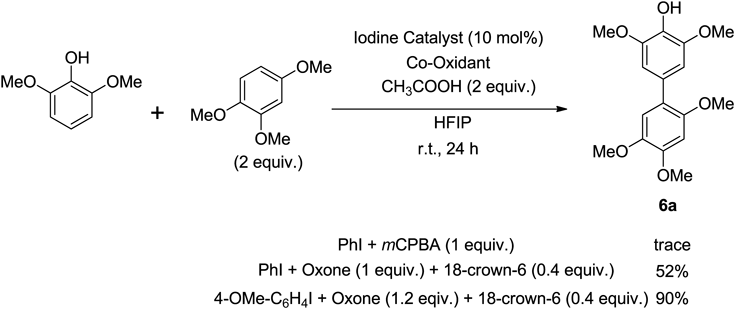
Recently we developed a stoichiometric hypervalent iodine(III) induced oxidative cross-coupling reaction of phenols.124) However, a catalytic version of the cross-coupling reaction of phenols using this hypervalent iodine(III) reagent remains to be achieved. Therefore, we investigated the oxidative cross-coupling reaction of phenols using an organic iodine(III) catalyst125) (Chart 15).
We chose a model reaction using 2,6-dimethoxyphenol and 1,2,4-trimethoxybenzene and a combination of iodoarene and mCPBA in HFIP. The use of mCPBA as the terminal oxidant was ineffective for phenol cross-coupling, as only trace amounts of cross-coupling products were obtained. To our delight, however, the use of Oxone led to the cross-coupled product in a yield of 52%. Various acids and solvents were carefully investigated in order to optimize the reaction conditions, and the best results were obtained when using 18-crown-6 and acetic acid (CH3COOH) and 4-methoxyiodobenzene as the iodine catalyst (Chart 16).
Under optimized reaction conditions, we next investigated the substrate scope to explore the versatility of the cross-coupling reaction between various phenols with arenes. As shown in Chart 17, cross-coupling product 6 was selectively obtained in good yield. Cross-coupling of electron-rich phenols afforded the corresponding cross-coupling products. The arenes with an amino group successfully reacted with 2,6-dimethoxyphenol to give the desired products 6b in good yields. The aniline derivatives gave the desired products 6c and 6d in moderate yields. The desired reaction proceeded in good yield bearing amino and methoxy groups, and gave the corresponding coupling products 6e–6g. The tosyl-protected indole also gave the corresponding coupling product 6h. When para-substituted phenol was used in this reaction, the coupling reaction occurred in the ortho-position. In the case of 4-methoxyphenol, the coupling reaction proceeded, and dimers 6i and 6j were obtained in good yields without the formation of overoxidized products. In the case of a coupling reaction of aniline and phenol, the reaction occurred in the para-position of the amino group. The steric effect of the amino groups was considered to be one reason for the regioselectivity of products 6m and 6n. The reaction also proceeded with halogen substituted naphthol to form the coupling product 6o.
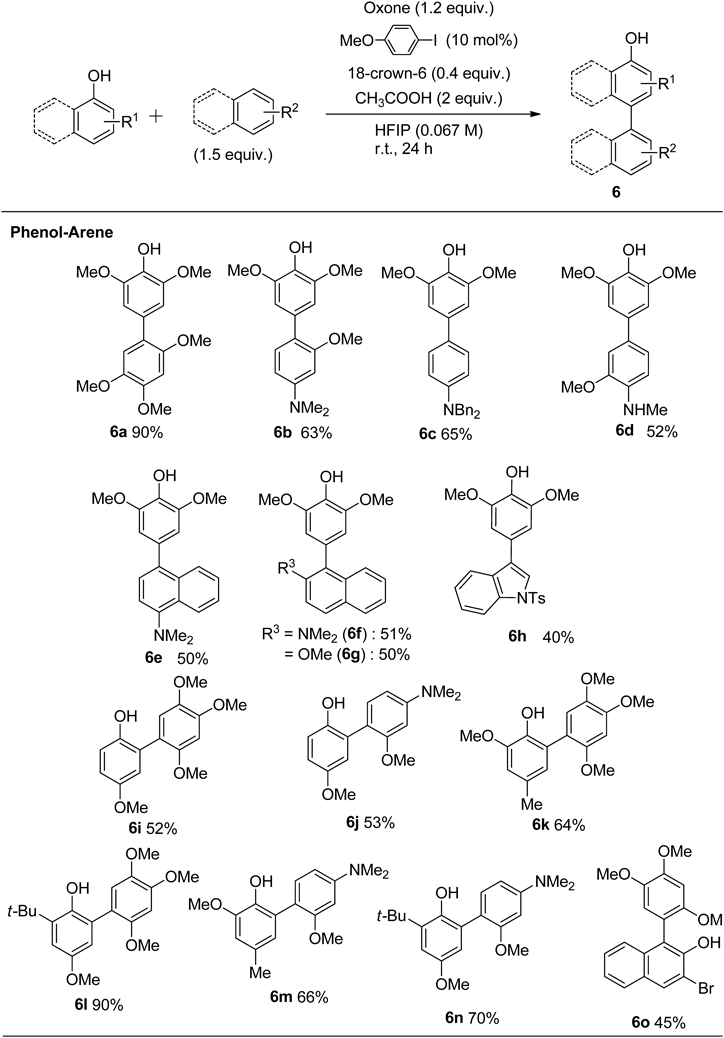
Further examination of the extent of the reaction showed that the reaction proceeded with two different phenols, with the coupling products 6p–6x obtained in good yield. Substrates bearing a bromo group were compatible with the reaction conditions, providing the corresponding products 6p and 6r in good yields. The coupling reaction proceeded with the trisubstituted phenol to produce biphenol 6s to 6u. The products (6v and 6w) were given in modest yields when 2,6-di-tert-butyl phenol was used. The 4-methylphenol gave the corresponding coupling product 6x in moderate yield (Chart 18).
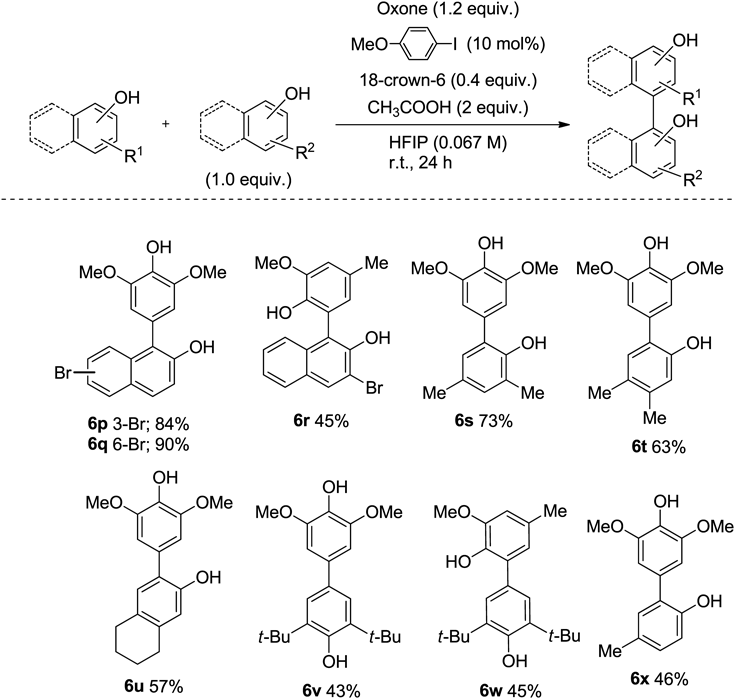
Our coupling method was applied to anilide cross coupling126) (Chart 19). The 2,2′-diiodobiphenyl catalyst was found to be effective in the coupling of aniline, giving the corresponding coupling products 7 in high yield.
The development of glycoscience revealed that complex oligosaccharides on the cell surface play an important role in biological events related to cell–cell adhesion, and medicines with glycosyl structures are now widely used.127–135) Therefore, the synthesis of biologically active oligosaccharides is important for organic chemists. Among them, thioglycosides have been most extensively studied in glycosylation reactions because of their stability and ease of preparation. However, glycosylation reactions of thioglycoside have historically required the use of highly toxic heavy metal oxidants.136–141) van Boom and colleagues have reported glycosylation reactions of thioglycosides using a combination of stoichiometric amounts of N-iodosuccinimide and catalytic amounts of triflic acid.142) Recently, we have reported a novel glycosylation reaction of a thioglycoside using hypervalent iodine(III) as an activator143,144) (Chart 20).
First, the readily available thioglycoside 8 as a donor and substrate 9 as an acceptor were treated with PIFA in dichloromethane in the presence of trifluoromethanesulfonic acid (TfOH). The corresponding glycosylated product 10a was obtained in moderate yield. Furthermore, when the reaction was performed with the donor 8b, which has a phthalimido group at the 2-position, the yield improved to 77% (Chart 21).
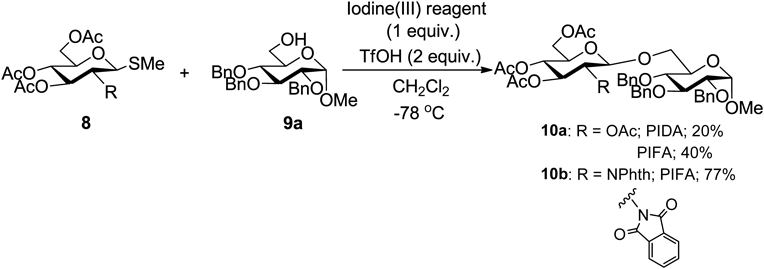
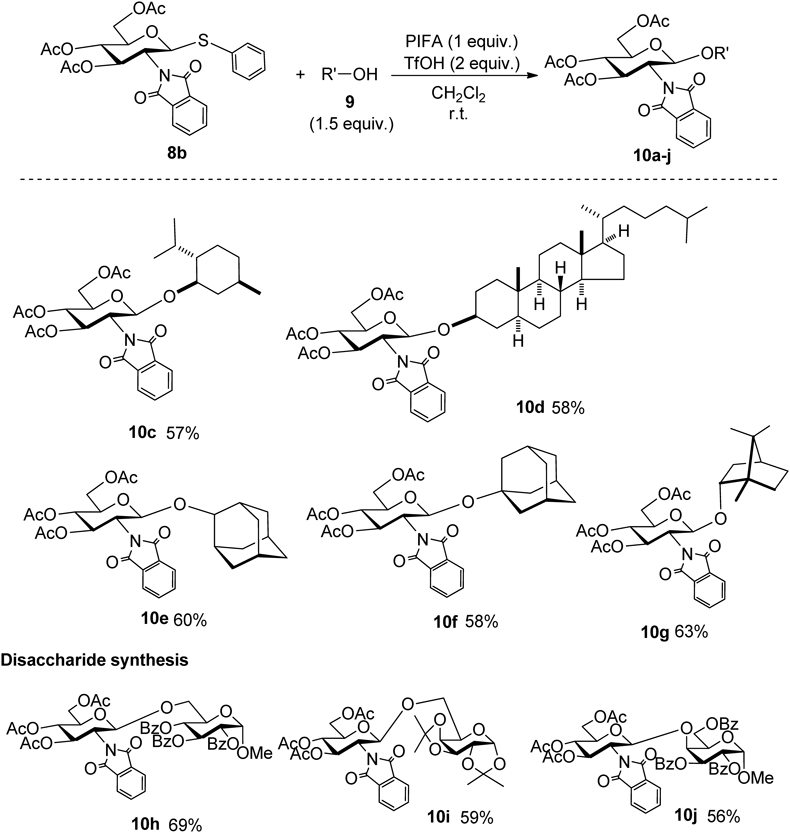
Under optimized reaction conditions, the glycosylation reaction proceeded with good yield (Chart 22). The reaction was carried out with an alkanol having a secondary alcohol on the rigid fused ring to give the corresponding glycosylation products 10c–10e. A sterically hindered tertiary alcohol, such as 1-adamantanol, also gave the product in 58% yield under the same reaction conditions. Moreover, a bulky secondary alcohol such as the (−)-borneol reacted with 8b to give the glycosylated product 10g in good yield. The synthesis of disaccharides with the above glycosylation method was also performed. When the benzoyl group-protected acceptor with a hydroxyl group at the 6-position was used, the corresponding glycosylated product 10h was obtained in high yield. A glycosylation reaction was carried out using an acetal-protected acceptor, and the reaction proceeded with good yield to obtain the desired product 10i. In addition, in the case of glycosylation with a galactose 4-hydroxyl group, the reaction proceeded with a yield of 56% to give the glycosylated product 10j.
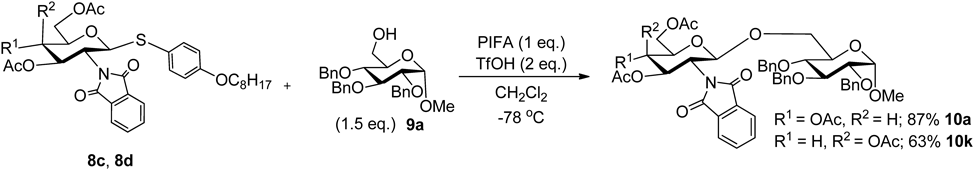
However, this method was developed based on the conventional method, which required an excess amount of odorous methanethiol or benzenethiol in the preparation of thioglycoside, and these thiols became a waste product during the reaction. Thus, we developed glycosylation using a thioglycoside donor, which was prepared with odorless p-octyloxybenzenethiol by the activation with the hypervalent iodine(III) reagent.145) The synthesis of disaccharides was perfomed using 1-O-methyl 2,3,4-tribenzyl-α-D-glucopyranoside 8c and 9a, respectively, as acceptor and donor substrates. The combination of “armed” and “disarmed” substrates appeared to be one of the most undesirable aspects of glycosylation. However, the reaction proceeded in 87% yield to obtain the desired disaccharide 10a (Chart 23). Based on the above results, the glycosylation reaction was examined using 8d as donor and 9a as the acceptor substrate.
We revealed that PIFA activates thioglycoside in the presence of TfOH and the glycosylation reaction proceeds. Furthermore, this method was useful for the synthesis of monosaccharides and disaccharides. Therefore, the combination of the thioglycoside and PIFA in the presence of TfOH provides an alternative method for the synthesis of biologically active oligosaccharides.
This report shows recent progress in metal-free biaryl synthesis using hypervalent iodine(III) as a reagent. The coupling reaction utilizes a low toxicity iodine reagent, and the desired product can be synthesized without functionalization of the substrates. The process is carried out under mild reaction conditions, is operationally simple, and allows the direct synthesis of various biaryls without functionalization. The novel methodology described herein is recognized as practical and extensible, and has demonstrated great potential for the synthesis of novel biaryls.
I would like to thank to my supervisor, Dr. Yasuyuki Kita, a professor at Ritsumeikan University. This work was partially supported by a Grant-in-Aid for Scientific Research (C), a Grant-in-Aid for Young Scientists (B), and a Grant-in-Aid for Research Activity Start-up from the JSPS.
The author declares no conflict of interest.