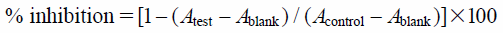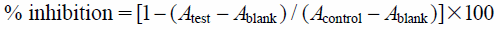Experimental
General Experimental ProceduresOptical rotations were measured on a JASCO P-2200 digital polarimeter. IR spectra were measured on JASCO FT/IR-6100 spectrophotometers. 1H- and 13C-NMR spectra were taken on a Bruker Avance III at 600 MHz and 150 MHz, respectively, with tetramethylsilane as an internal standard. CD spectra were obtained with a JASCO J-720 spectropolarimeter. Positive and negative-ion HR-ESI-MS were performed with a Thermo Fisher Scientific LTQ Orbitrap XL. Silica gel column chromatography (CC) was performed on silica gel 60 (70–230 mesh) (E. Merck, Darmstadt, Germany) and reversed-phase octadecylsilanized (ODS) open CC on Cosmosil 75C18-OPN (Nacalai Tesque, Kyoto, Japan) (Φ = 50 mm, L = 25 cm). HPLC was performed on an ODS column [Inertsil ODS-3 (GL Science Inc., Tokyo, Japan; Φ = 10 mm, L = 25 cm, 4.0 mL/min), Cosmosil πNAP (Nacalai Tesque; Φ = 10 mm, L = 25 cm, 4.0 mL/min) and Cosmosil PBr (Nacalai Tesque; Φ = 10 mm, L = 25 cm, 4.0 mL/min)], and the eluate was monitored with photodiode arrtay (200–400 nm) and refractive index monitors. Crude β-glucosidase (Sumizyme BGA) was a generous gift from Shin Nihon Chemical Co., Ltd. (Anjo, Aichi, Japan) (Lot No. 0930708-03). Crude naringinase was from Amano Enzyme Inc. (Nagoya, Aichi, Japan) as a gift (Lot No. NAG1252306). MTPAs were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan).
Plant MaterialLeaves of D. maritima were collected in Taketomi-cho, Yaeyama-gun, Okinawa, Japan, in November, 2003 and a voucher specimen was deposited in the Herbarium of Pharmaceutical Sciences, Graduate School of Biomedical and Health Sciences, Hiroshima University (03-DM-Okinawa-1105). The plant was identified by one of the authors (M.A.).
Extraction and IsolationAir-dried leaves of D. maritima (7.80 kg) were extracted with MeOH (45 L) three times. The MeOH extract was concentrated to 6 L and then washed with n-hexane (6 L, 245 g). The methanolic layer was concentrated to a viscous gum. The gummy mass was suspended in H2O (6 L), and then partitioned with ethyl acetate (EtOAc) (6 L) and 1-butanol (1-BuOH) (6 L), successively, to give 397 g and 216 g of EtOAc and 1-BuOH-soluble fractions. The remaining water-layer was concentrated to give a H2O-soluble fraction (245 g). The 1-BuOH-soluble fraction was subjected to a Diaion HP-20 CC (Φ = 80 mm, L = 50 cm), and eluted with H2O–MeOH (4 : 1, 5 L), (3 : 2, 5 L), (2 : 3, 5 L), and (1 : 4, 5 L), and MeOH (5 L), 1 L-fractions being collected.
The residue (17.5 g) in fractions 4–7 of a Diaion HP-20 CC was subjected to silica gel CC (Φ = 40 mm, L = 55 cm), and eluted with CHCl3 (3 L), CHCl3–MeOH (99 : 1, 3 L), (49 : 1, 3 L), (97 : 3, 3 L), (19 : 1, 3 L), (37 : 3, 3 L), (9 : 1, 3 L), (7 : 1, 3 L), (17 : 3, 3 L), (33 : 7, 3 L), (4 : 1, 3 L), (3 : 1, 3 L), and (7 : 3, 3 L), 500 mL-fractions being collected. Compounds 6 (266 mg) and 5 (437 mg) were obtained in fractions 18–19 and 23–26, respectively.
The residue (29.2 g) in fractions 11–14 of a Diaion HP-20 CC was subjected to silica gel CC (Φ = 50 mm, L = 54.5 cm), and eluted with CHCl3 (3 L), CHCl3–MeOH (99 : 1, 3 L), (49 : 1, 3 L), (97 : 3, 3 L), (19 : 1, 3 L), (37 : 3, 3 L), (9 : 1, 3 L), (7 : 1, 3 L), (17 : 3, 3 L), (33 : 7, 3 L), (4 : 1, 3 L), (3 : 1, 3 L), and (7 : 3, 3 L), 500 mL-fractions being collected. The residue (3.00 g out of 6.74 g) in fractions 57–65 of silica gel CC was separated by ODS CC (Φ = 50 mm, L = 25 cm), and eluted with a linear gradient solvent system from MeOH–H2O (1 : 9, 2 L) to MeOH–H2O (9 : 1, 2 L), 10 g-fractions being collected. The residue (160 mg) in fractions 223–234 was purified by HPLC (Inertsil ODS-3, H2O–MeOH, 1 : 1) to give 36.4 mg of 3 from the peak at 10.2 min. The residue (88.1 mg) in fractions 240–249 was purified by HPLC (Cosmosil πNAP, H2O–MeOH, 2 : 3) to give 32.8 mg of 4 from the peak at 10.2 min. The residue (2.81 g) in fractions 66–71 of silica gel CC was separated by ODS CC (Φ = 50 mm, L = 25 cm), and eluted with a linear gradient solvent system from MeOH–H2O (1 : 9, 2 L) to MeOH–H2O (9 : 1, 2 L), 10 g-fractions being collected. The residue (136 mg) in fractions 106–129 was purified by HPLC (Cosmosil PBr, H2O–MeOH, 3 : 2) to give 15.0 mg of 7 and 19.9 mg of 8 from the peaks at 25.0 min and 26.4 min, respectively.
The residue (64.5 g) in fractions 5–19 of a Diaion HP-20 CC was subjected to silica gel CC (Φ = 80 mm, L = 40 cm), and eluted with CHCl3 (6 L), CHCl3–MeOH (99 : 1, 6 L), (49 : 1, 6 L), (97 : 3, 6 L), (19 : 1, 6 L), (37 : 3, 6 L), (9 : 1, 6 L), (7 : 1, 6 L), (17 : 3, 6 L), (33 : 7, 6 L), (4 : 1, 6 L), (3 : 1, 6 L), and (7 : 3, 6 L), 1 L-fractions being collected. The residue (3.00 g out of 12.3 g) in fractions 53–62 was separated by ODS CC (Φ = 50 mm, L = 25 cm), and eluted with a linear gradient solvent system from MeOH–H2O (1 : 9, 2 L) to MeOH–H2O (9 : 1, 2 L), 10 g-fractions being collected. The residue (63.0 mg) in fractions 243–246 was purified by HPLC (Inertsil ODS-3, H2O–MeOH, 3 : 7) to give 10.8 mg of 1 from the peak at 5.9 min. The residue (196 mg) in fractions 247–263 was purified by HPLC (Inertsil ODS-3, H2O–MeOH–CH3COOH, 7 : 13 : 0.1) to give 3.8 mg of 2 from the peak at 18.4 min.
Ebenamarioside A (1)Colorless amorphous powder, [α]D25 +15.1 (c = 0.72, MeOH); IR νmax (film) cm−1: 3439, 2928, 2871, 1745, 1636, 1458, 1072; 1H-NMR (600 MHz, pyridine-d5): Table 1; 13C-NMR (150 MHz, pyridine-d5): Table 2; HR-ESI-MS (positive-ion mode): m/z: 819.4499 [M + Na]+ (Calcd for C42H68O14Na: 819.4501).
Ebenamarioside B (2)Colorless amorphous powder, [α]D27 −5.6 (c = 0.43, MeOH); IR νmax (film) cm−1: 3393, 2936, 2872, 1686, 1043; 1H-NMR (600 MHz, pyridine-d5): Table 1; 13C-NMR (150 MHz, pyridine-d5): Table 2; HR-ESI-MS (negative-ion mode): m/z: 795.4529 [M − H]− (Calcd for C42H67O14: 795.4525).
Ebenamarioside C (3)Colorless amorphous powder, [α]D27 +14.0 (c = 0.10, pyridine); IR νmax (film) cm−1: 3353, 2927, 2876, 1705, 1045; 1H-NMR (600 MHz, pyridine-d5): Table 1; 13C-NMR (150 MHz, pyridine-d5): Table 2; HR-ESI-MS (positive-ion mode): m/z: 835.4447 [M + Na]+ (Calcd for C42H68O15Na: 835.4450).
Ebenamarioside D (4)Colorless amorphous powder, [α]D26 −5.6 (c = 0.86, MeOH); IR νmax (film) cm−1: 3400, 2932, 2877, 1740, 1071; 1H-NMR (600 MHz, pyridine-d5): Table 1; 13C-NMR (150 MHz, pyridine-d5): Table 2; HR-ESI-MS (positive-ion mode): m/z: 835.4449 [M + Na]+ (Calcd C42H68O15Na: 835.4450).
Compound 5Colorless amorphous powder, [α]D26 +10.2 (c = 0.33, MeOH); IR νmax (film) cm−1: 3379, 2930, 2876, 1450, 1375, 1135, 1043; 1H-NMR (600 MHz, CD3OD): Table 3; 13C-NMR (150 MHz, CD3OD): Table 3; HR-ESI-MS (positive-ion mode): m/z: 265.1411 [M + Na]+ (Calcd C13H22O4Na: 265.1410).
Compound 6Colorless syrup, [α]D26 approx. 0.00 (c = 0.81, MeOH); IR νmax (film) cm−1: 3414, 2932, 2877, 1715, 1455, 1242, 1042; 1H-NMR (600 MHz, CD3OD): Table 3; (CDCl3) δ: 6.22 (1H, dd, J = 15.4, 5.4 Hz, H-8), 5.91 (1H, dd, J = 15.4, 1.2 Hz, H-7), 4.45 (1H, qd, J = 6.4, 5.4 Hz, H-9), 3.91 (1H, dd, J = 8.3, 3.0 Hz, H-12a), 3.75 (1H, d, J = 8.3 Hz, H-12b), 2.65 (1H, d, J = 18.3 Hz, H-4a), 2.60 (1H, dd, J = 18.3, 2.0 Hz. H-4b), 2.55 (1H, dd, J = 18.1, 3.0 Hz, H-2a), 2.42 (1H, dd, J = 18.1, 2.0 Hz, H-2b), 1.33 (3H, d, J = 6.4 Hz, H3-10), 1.21 (3H, s, H3-13); 0.99 (3H, s, H3-11); 13C-NMR (150 MHz, CD3OD): Table 3; (CDCl3) δ: 208.6 (C-3), 139.8 (C-8), 123.7 (C-7), 85.5 (C-5), 81.6 (C-6), 77.2 (C-12), 68.1 (C-9), 52.6 (C-4), 52.5 (C-2), 47.7 (C-1), 24.0 (C-10), 18.7 (C-13), 15.6 (C-11); CD (c = 6.46 × 10−5 M, MeOH) ∆ε (nm): +0.64 (241), −0.30 (296); HR-ESI-MS (positive-ion mode): m/z: 263.1254 [M + Na]+ (Calcd C13H20O4Na: 263.1254).
Sugar AnalysisAbout 500 µg each of 1–4 was hydrolyzed with 1 M HCl (0.1 mL) at 90°C for 2 h. The reaction mixtures were partitioned with an equal amount of EtOAc (0.1 mL), and the water layers were analyzed by HPLC with a chiral detector (JASCO OR-4090) on an amino column [InertSustain NH2, 4.6 × 250 mm (GL Science Inc.), CH3CN–H2O (4 : 1), flow rate: 1 mL/min]. All the hydrolyzates gave a peak for D-glucose at 10.9 min with positive optical rotation signs. The peaks were identified by co-chromatography with an authentic sample.
Enzymatic Hydrolysis of 1, 3 and 4 to 1a, 1b and 1c, 3a, 3b and 3c, and 4a, 4b and 4c, RespectivelyEbenamarioside A (1) (9.8 mg) in 1 mL of H2O hydrolyzed with 15 mg of crude glucosidase at 37°C for 72 h. The reaction mixture was subjected to silica gel CC (Φ = 2 cm, L = 15 cm) with increasing amounts of MeOH in CHCl3 [CHCl3–MeOH (9 : 1, 50 mL), (9 : 1, 100 mL to 7 : 3, 100 mL, linear gradient), (7 : 3, 50 mL) and (1 : 1, 100 mL)], 10-mL fractions being collected. An aglycone (1c) (2.8 mg) was obtained in fractions 5–6 and the monosaccharide mixture (1a and 1b) (5.5 mg) in fractions 9–12. The mixture fraction was purified by HPLC (ODS-3, H2O–MeOH, 4 : 1) to give 1.9 mg of 1a and 3.0 mg of 1b from the peaks at 4.8 min and 8.7 min, respectively. Mesembryanthenoidigenic acid 28-O-β-D-glucopyranosyl ester (1a): Amorphous powder, [α]D25 +31.4 (c = 0.09, MeOH), HR-ESI-MS (positive-ion mode) m/z: 657.3975 [M + Na]+ (Calcd for C36H58O9Na: 657.3973)9); mesembryanthenoidigenic acid 30-O-β-D-glucopyranoside (1b): Amorphous powder, [α]D25 +14.5 (c = 0.15, MeOH), IR νmax (film) cm−1: 3370, 2929, 2867, 1686, 1636, 1457, 1077; 1H-NMR (600 MHz, pyridine-d5) δ: 5.47 (1H, dd, J = 3.3, 3.3 Hz, H-12), 4.61 (1H, dd, J = 11.7, 2.1 Hz, H-6″a), 4.44 (1H, dd, J = 11.7, 5.3 Hz, H-6″b), 4.87 (1H, d, J = 7.7 Hz, H-1″), 4.28 (1H, dd, J = 8.7, 8.4 Hz, H-4″), 4.27 (1H, dd, J = 8.4, 8.3 Hz, H-3″), 4.10 (1H, dd, J = 8.3, 7.7 Hz, H-2″), 4.00 (1H, m, H-5″), 3.95 (1H, d, J = 9.3 Hz, H-29a), 3.46 (1H, dd, J = 10.2, 5.6 Hz, H-3), 3.44 (1H, d, J = 9.3 Hz, H-29b), 3.37 (1h, dd, J = 13.6, 3.9 Hz, H-18), 2.19 (1H, ddd, J = 13.2, 13.0, 3.6 Hz, H-15a), 2.12 (1H, ddd, J = 13.0, 13.0, 3.0 Hz, H-16a), 2.08 (1H, ddd, J = 14.0, 14.0, 4.0 Hz, H-22a), 2.04 (1H, dd, J = 13.9, 13.8 Hz, H-19a), 1.93 (3H, m, H2-11 and H-16b), 1.84 (3H, m, H2-2 and H-22b), 1.75 (1H, ddd, J = 13.6, 13.3, 3.6 Hz, H-21a), 1.66 (1H, dd, J = 11.0, 6.7 Hz, H-9), 1.57 (1H, m. H-6a), 1.56 (1H, m, H-1a), 1.51 (1H, ddd, J = 12.6, 12.6, 3.5 Hz, H-7a), 1.45 (1H, dd, J = 13.6, 4.2 Hz, H-19b), 1.43 (1H, m, H-21b), 1.38 (1H, m, H-6b), 1.33 (1H, m, H-7b), 1.26 (3H, s, H3-27), 1.25 (3H, s, H3-23), 1.21 (3H, s, H3-30), 1.18 (1H, m, H-15b), 1.04 (3H, s, H3-24), 1.03 (3H, s, H3-26), 1.01 (1H, m, H-1b), 0.91 (3H, s, H3-25), 0.87 (1H, br d, J = 10.4 Hz, H-5); 13C-NMR (150 MHz, pyridine-d5): Table 2, HR-ESI-MS (positive-ion mode) m/z: 633.4000 [M − H]− (Calcd for C36H57O9: 633.3997); mesembryanthenoidigenic acid (1c): [α]D25 +24.9 (c = 0.14, MeOH), HR-ESI-MS (positive-ion mode) m/z: 471.3473 [M − H]− (Calcd for C30H47O4: 471.3469).10,11)
Ebenamarioside C (3) (9.5 mg) in 1 mL of H2O was hydrolyzed with crude naringinase (15 mg) at 37°C for 72 h. The reaction mixture was subjected silica gel CC (Φ = 2 cm, L = 15 cm) with increasing amounts of MeOH in CHCl3 [CHCl3-MeOH (9 : 1, 50 mL), (9 : 1, 100 mL to 7 : 3, 100 mL, linear gradient), (7 : 3, 50 mL) and (1 : 1, 100 mL)], 10-mL fractions being collected. 29-O-β-D-glucopyranosyl estre (3a) was obtained in fractions 11–13. Similarly ebenamarioside C (3) (15 mg) in 1 mL of H2O was hydrolyzed with crude β-glucosidase and silica gel CC with the same condition as above gave 5.2 mg of an aglycone (3c) and 5.6 mg of 29-O-β-D-glucopyranoside (3b) in fractions 5–8 and 11–13, respectively. 2α,3β,29-Trihydroxyolean-12-en-28-oic acid 28-O-β-D-glucopyranosyl ester (3a): Amorphous powder, [α]D24 +18.7 (c = 0.08, MeOH), 13C-NMR (150 MHz, pyridine-d5, MeOH-d4 and DMSO-d6): Table 1, HR-ESI-MS (positive-ion mode) m/z: 673.3922 [M + Na]+ (Calcd for C36H58O10Na: 673.3922). 2α,3β,29-Trihydroxyolean-12-en-28-oic acid 29-O-β-D-glucopyranoside (3b): Amorphous powder; [α]D24 +9.6 (c = 0.28, MeOH); IR νmax (film) cm−1: 3379, 2934, 2872, 1687, 1459, 1050; 1H-NMR (600 MHz, pyridine-d5) δ: 5.43 (1H, dd, J = 3.3, 3.3 Hz, H-12), 4.87 (1H, d, J = 7.7 Hz, H-1″), 4.60 (1H, dd, J = 11.8, 2.2 Hz, H-6″a), 4.44 (1H, dd, J = 11.8, 5.3 Hz, H-6″b), 4.28 (2H, m, H-3″ and 4″), 4.10 (2H, m, H-2 and 2″), 4.00 (1H, m, H-5″), 3.94 (1H, d, J = 9.1 Hz, H-29a), 3.43 (1H, d, J = 9.1 Hz, H-29b), 3.40 (1H, d, J = 9.6 Hz, H-3), 3.35 (1H, dd, J = 13.3, 3.1 Hz, H-18), 2.25 (1H, dd, J = 12.4, 4.2 Hz, H-1a), 2.18 (1H, ddd, J = 13.3, 13.0, 3.1 Hz, H-15a), 2.10 (1H, m, H-11a), 2.05 (1H, m, H-22a), 2.01 (1H, m, H-19a), 1.98 (2H, m, H2-16), 1.95 (1H, m, H-11b), 1.84 (1H, br d, J = 13.6 Hz, H-22b), 1.76 (1H, m, H-9), 1.75 (1H, m, H-21a), 1.56 (1H, m, H-6a), 1.51 (1H, m, H-7a), 1.43 (2H, m, H-19b and 21b), 1.38 (1H, m, H-6b), 1.31 (1H, m, H-7b), 1.28 (1H, m, H-1b), 1.27 (3H, s, H3-23), 1.23 (3H, s, H3-27), 1.21 (3H, s, H3-30), 1.20 (1H, m, H-15b), 1.08 (3H, s, H3-24), 1.02 (3H, s, H3-26), 1.01 (1H, m, H-5), 0.99 (3H, s, H3-25, 13C-NMR (150 MHz, pyridine-d5): Table 2; HR-ESI-MS (positive-ion mode) m/z: 649.3951 [M − H]− (Calcd for C36H57O10: 649.3946). 2α,3β,29-Trihydroxyolean-12-en-28-oic acid (3c): Amorphous powder; [α]D24 +34.6 (c = 0.26, MeOH); HR-ESI-MS (positive-ion mode) m/z: 487.3422 [M − H]− (Calcd for C30H47O5: 487.3418).
Ebenamarioside C (4) (16.6 mg) in 1 mL of H2O was hydrolyzed with crude β-glucosidase 37°C for 72 h. The reaction mixture was separated by silica gel CC with the same condition as above to give 4.1 mg of an aglycone (4c) and 9.4 mg of a mixture of two monoglucosidic compounds (4a and 4b) in fractions 5–8 and 10–15, respectively. The mixture was purified by HPLC (ODS-3, H2O–MeOH, 4 : 1) to afford 6.3 mg of 28-O-β-D-glucopyranosyl ester (4a) and 1.9 mg of 30-O-β-D-glucopyranoside (4b) from the peaks at 3.9 min and 9.2 min, respectively. 2α,3β,30-Trihydroxyurs-12-en-28-oic acid 28-O-β-D-glucopyranosyl ester (4a): Amorphous powder; [α]D24 +17.9 (c 0.31, MeOH); IR νmax (film) cm−1: 3373, 2925, 2877, 1732, 1456, 1073; 1H-NMR (600 MHz, pyridine-d5) δ: 6.31 (1H, d, J = 8.1 Hz, H-1′), 5.48 (1H, dd, J = 3.4, 3.4 Hz, H-12), 4.47 (1H, dd, J = 11.8, 2.4 Hz, H-6′a), 4.41 (1H, dd, J = 11.8, 4.4 Hz, H-6′b), 4.39 (1H, dd, J = 9.5, 8.9 Hz, H-4′), 4.31 (1H, dd, J = 8.9, 8.7 Hz, H-3′), 4.23 (1H, dd, J = 8.7, 8.1 Hz, H-2′), 4.11 (1H, ddd, J = 11.1, 9.6, 4.3 Hz, H-2), 4.04 (1H, ddd, J = 9.5, 4.4, 2.4 Hz, H-5′), 3.93 (1H, dd, J = 10.7, 2.9 Hz, H-30a), 3.88 (1H, dd, J = 10.7, 5.6 Hz, H-30b), 3.39 (1H, d, J = 9.4 Hz, H-3), 2.65 (1H, d, J = 11.5 Hz, H-18), 2.49 (1H, ddd, J = 13.8, 13.7, 4.4 Hz, H-15a), 2.26 (1H, dd, J = 12.5, 4.4 Hz, H-1a), 2.21 (1H, ddd, J = 13.5, 13.4, 4.2 Hz, H-16a), 2.07 (1H, m, H-22a), 2.06 (1H, m, H-16b), 2.05 (2H, m, H2-11), 2.01 (1H, m, H-19), 1.86 (2H, m, H2-21), 1.84 (1H, m, H-22b), 1.74 (1H, dd, J = 10.0, 7.4 Hz, H-9), 1.54 (1H, m, H-7a), 1.52 (1H, m, H-6a), 1.40 (1H, m, H-7b), 1.38 (1H, m, H-6b), 1.27 (1H, m, H-1b), 1.26 (3H, s, H3-23), 1.20 (3H, s, H3-27), 1.19 (3H, s, H3-26), 1.18 (1H, m, H-15b), 1.16 (1H, m, H-20), 1.11 (3H, d, J = 6.4 Hz, H3-29), 1.09 (3H, s, H3-24), 1.04 (3H, s, H3-25), 1.01 (1H, d, J = 12.1 Hz, H-5); 13C-NMR (150 MHz, pyridine-d5): Table 2; HR-ESI-MS (positive-ion mode) m/z: 673.3925 [M + Na]+ (Calcd for C36H58O10Na: 673.3922).
2α,3β,30-Trihydroxyurs-12-en-28-oic acid 30-O-β-D-glucopyranoside (4b): Amorphous powder; [α]D25 +5.5 (c = 0.10, MeOH); IR νmax (film) cm−1: 3370, 2930, 2871, 1686, 1457, 1050; 1H-NMR (600 MHz, pyridine-d5) δ: 5.45 (1H, dd, J = 3.1, 3.1 Hz, H-12), 4.90 (1H, d, J = 7.8 Hz, H-1″), 4.62 (1H, dd, J = 11.7, 2.2 Hz, H-6″a), 4.45 (1H, dd, J = 11.7, 5.3 Hz, H-6″b), 4.30 (1H, dd, J = 8.8, 8.9 Hz, H-3″), 4.27 (1H, dd, J = 8.9, 8.9 Hz, H-4″), 4.10 (1H, m, H-2), 4.09 (1H, m, H-2″), 4.07 (1H, m, H-30a), 4.03 (1H, m, H-5″), 3.89 (1H, dd, J = 9.5, 3.5 Hz, H-30b), 3.40 (1H, d, J = 9.4 Hz, H-3), 2.65 (1H, d, J = 11.3 Hz, H-18), 2.32 (1H, ddd, J = 13.8, 13.8, 4.4 Hz, H-15a), 2.24 (1H, dd, J = 12.5, 4.4 Hz, H-1a), 2.07 (1H, m, H-16a), 2.04 (1H, m, H-21a), 2.03 (1H, m, H-22a), 2.02 (2H, m, H2-11), 1.96 (1H, m, H-16b), 1.94 (1H, m, H-22b), 1.75 (2H, m, H-9 and 19), 1.66 (1H, m, H-21b), 1.55 (1H, m, H-6a), 1.54 (1H, m, H-7a), 1.39 (1H, m, H-20), 1.38 (1H, m, H-6b), 1.37 (1H, m, H-7b), 1.28 (3H, s, H3-23), 1.28 (1H, m, H-1b), 1.17 (3H, s, H3-27), 1.16 (1H, m, H-15b), 1.08 (3H, s, H3-24), 1.04 (3H, s, H3-26), 1.03 (1H, m, H-5), 1.01 (3H, d, J = 6.4 Hz, H3-29), 0.98 (3H, s, H3-25); 13C-NMR (150 MHz, pyridine-d5): Table 2; HR-ESI-MS (positive-ion mode) m/z: 649.3948 [M − Na]− (Calcd for C30H57O10: 649.3946).
2α,3β,30-Trihydroxyurs-12-en-28-oic acid (4c): Amorphous powder; [α]D23 +17.7 (c = 0.13, EtOH); HR-ESI-MS (positive-ion mode) m/z: 487.3423 [M − H]− (Calcd for C30H47O5: 487.3418).
Preparation of (R)- and (S)-MTPA Esters (5a and 5b) from 5A solution of 5 (1.0 mg) in 0.5 mL of dry CH2Cl2 were reacted with (R)-MTPA (21.5 mg) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (13.4 mg) and N,N-dimethyl-4-aminopyridine (4-DMAP) (16.1 mg). The mixture was then occasionally stirred at 37°C for 24 h. After the addition of CHCl3 (1.5 mL), the reaction mixture was successively washed with H2O (1 mL), 1 M HCl (1 mL), NaHCO3-saturated H2O (1 mL), and brine (1 mL). The organic layer was dried with Na2SO4 and evaporated under reduced pressure. The residue was purified by preparative TLC [silica gel (0.25 mm thickness), being applied for 8 cm width, with development with CHCl3–MeOH (19 : 1) for 9 cm and then eluting with CHCl3–MeOH (1 : 1)] to furnish an ester 5a (0.5 mg) from the band at Rf = 0.67. Through the same procedure, 5b (0.6 mg, Rf = 0.59) were prepared from 5 (1.0 mg) using (S)-MTPA (25.4 mg), EDC (15.8 mg), and 4-DMAP (15.5 mg), respectively.
(R)-MTPA ester of 5 (5a): amorphous powder; 1H-NMR (600 MHz, CDCl3) δ: 7.35–7.55 (5H, m, aromatic protons), 6.05 (1H, d, J = 15.2 Hz, H-7), 6.02 (1H, dd, J = 15.2, 4.9 Hz, H-8), 5.66 (1H, qd, J = 6.5, 4.9 Hz, H-9), 5.44 (1H, dddd, J = 10.5, 10.5, 7.2, 7.2 Hz, H-3), 3.87 (1H, d, J = 8.2 Hz, H-12a), 3.77 (1H, dd, J = 8.2, 2.0 Hz, H-12b), 3.53 (3H, s, –OMe), 3.51 (3H, s, –OMe), 2.18 (1H, ddd, J = 13.6, 7.2, 1.5 Hz, H-4), 2.03 (1H, ddd, J = 13.6, 7.2, 1.5 Hz, H-2), 1.73 (1H, dd, J = 13.6, 10.5 Hz, H-4), 1.70 (1H, ddd, J = 13.6, 10.5, 2.0 Hz, H-2), 1.45 (3H, d, J = 6.5 Hz, H3-10), 1.08 (3H, s, H3-13), 0.90 (3H, s, H3-11); HR-ESI-MS (positive-ion mode) m/z: 697.2206 [M + Na]+ (Calcd for C33H36O8F6Na: 697.2207).
(S)-MTPA ester of 5 (5b): amorphous powder; 1H-NMR (600 MHz, CDCl3) δ: 7.35–7.55 (10H, m, aromatic protons), 6.11 (1H, d, J = 15.5 Hz, H-7), 6.08 (1H, dd, J = 15.5, 5.5 Hz, H-8), 5.63 (1H, qd, J = 6.5, 5.5 Hz, H-9), 5.44 (1H, dddd, J = 10.7, 10.7, 7.1, 7.1 Hz, H-3), 3.88 (1H, d, J = 8.3 Hz, H-12a), 3.78 (1H, dd, J = 8.3, 2.1 Hz, H-11b), 3.52 (3H, s, –OMe), 3.49 (3H, s, –OMe), 2.25 (1H, ddd, J = 13.6, 7.1, 1.5 Hz, H-4), 1.99 (1H, ddd, J = 13.6, 7.1, 1.5 Hz, H-2), 1.85 (1H, ddd, J = 13.6, 10.7 Hz, H-4), 1.62 (1H, ddd, J = 13.6, 10.7, 2.1 Hz, H-2), 1.40 (3H, d, J = 6.5 Hz, H3-10), 1.10 (3H, s, H3-13), 0.91 (3H, s, H3-11); HR-ESI-MS (positive-ion mode) m/z: 697.2206 [M + Na]+ (Calcd for C33H36O8F6Na: 697.2207).
NaBH4 Reduction of 6To a solution of 6 (11.0 mg) in MeOH (1 mL) was added 8.2 mg of NaBH4 and the reaction mixture was stirred for 5 min at 25°C. Excess NaBH4 was quenched by the addition of 1 mL of acetone and then the reaction mixture was evaporated to dryness. The resultant residue was purified by HPLC [Inertsil ODS-3, 6 × 250 mm, H2O–MeOH (1 : 4), flow rate: 1.6 mL/min] to give 3.1 mg of 6c (=5) and 5.8 mg of 6d from the peaks at 10.9 min and 13.3 min. respectively.
Compound 6c: amorphous powder; [α]D24 +7.1 (c = 0.31, MeOH); HR-ESI-MS (positive-ion mode) m/z: 265.1409 [M + Na]+ (Calcd for C13H22O4Na: 265.1410).
Compound 6d: amorphous powder; [α]D24 −5.5 (c = 0.29, MeOH); IR νmax (film) cm−1: 3402, 2929, 2889, 1604, 1453, 1381, 1101, 1053; 1H-NMR (600 MHz, CD3OD) δ: 6.02 (1H, dd, J = 15.5, 5.6 Hz, H-8), 5.82 (1H, dd, J = 15.5, 1.0 Hz, H-7), 4.33 (1H, qd, J = 6.4, 5.6 Hz, H-9), 4.12 (1H, d, J = 6.9 Hz, H-12a), 4.02 (1H, dd, J = 5.7, 5.7 Hz, H-3), 3.76 (1H, dd, J = 6.9, 2.3 Hz, H-12b), 2.13 (1H, dd, J = 15.5, 5.7 Hz, H-4a), 2.02 (1H, ddd, J = 15.3, 5.7, 2.2 Hz, H-2a), 1.84 (1H, dd, J = 15.5, 2.2 Hz, H-4b), 1.73 (1H, dd, J = 15.3, 2.3 Hz, H-2b), 1.25 (3H, d, J = 6.4 Hz, H3-10), 1.13 (3H, s, H3-13), 0.86 (3H, s, H3-11); 13C-NMR (150 MHz, CD3OD) δ: 139.4 (C-8), 126.8 (C-7), 87.2 (C-5), 82.5 (C-6), 76.3 (C-12), 69.1 (C-9), 66.0 (C-3), 48.0 (C-1), 45.1 (C-4), 44.9 (C-2), 24.1 (C-10), 19.7 (C-13), 16.3 (C-11); HR-ESI-MS (positive-ion mode) m/z: 265.1408 [M + Na]+ (Calcd for C13H22O4Na: 265.1410).
Cytotoxic Activity toward Human Lung Adenocarcinoma, A549 CellsCytotoxic activity toward lung adenocarcinoma cells was determined by colorimetric cell viability assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Lung adenocarcinoma cell line A549 was purchased from the JCRB Cell Bank, Japan. A549 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat inactivated FCS, and kanamycin (100 µg/mL) and amphotericin B (5.6 µg/mL). In a 96-well plate, 1 µL aliquots of sample solutions and the cancer cells (5 × 103 cells/well) in 100 µL medium were added to each well, and then the plate incubated at 37°C under a 5% CO2 atmosphere for 72 h. A solution (100 µL) of MTT (0.5 mg/mL) was then added to each well and the incubation was continued for a further 1 h. The absorbance of each well was measured at 540 nm using a Molecular Devices Versamax tunable microplate reader. DMSO was used as a negative control and doxorubicin as a positive control. The cytotoxic activity was calculated as:
where Acontrol is the absorbance of the control DMSO well, Atest the absorbance of the test wells, and Ablank the absorbance of the cell-free wells.
Anti-Leishmania ActivityThe anti-Leishmania major activity toward promastigotes was determined by the colorimetric cell viability MTT assay. The promastigotes at the logarithmic growth phase were cultured in M199 medium supplemented with 10% heat-inactivated fetal bovine serum and 100 µg/mL of kanamycin. In a 96-well plate, 1 µL aliquot of sample solutions and L. major cells (1 × 105 cells/well) in 100 µL medium were added to each well, and then the plate was incubated at 27°C under an ambient atmosphere for 72 h. A solution of MTT (100 µL) was then added to each well and the incubation was continued overnight. The formazan product of MTT reduction was then dissolved in DMSO and then the absorbance was measured using a Molecular Devices Versamax tunable microplate reader. DMSO was used as a negative control and amphotericin B as a positive control. The experiment was performed in triplicate. The anti-Leishmania major activity was quantified as the percentage of the control absorbance of reduced dye at 540 nm. The inhibitory activity was calculated as:
where Acontrol is the absorbance of the control (DMSO) well, Atest the absorbance of the test wells, and Ablank the absorbance of the cell-free wells.