Aromatic compounds having a thiocyanate unit have been used as a substructure of bioactive molecules and building blocks in organic synthesis.1) For example, phenols, anilides and heteroaromatic compounds with a thiocyanate group are known to show herbicidal acitivity,2) hypolipidemic activity,3) anti-cancer activity,4) antifungal activity,5) and antimicrobial activity.6,7) Aryl thiocyanate unit can be not only transformed to sulfide,8–10) disulfide,11) tetrazole,12) and thiocarbamate13,14) but also act as cyanate and/or thiolate sources in cross coupling reactions and difunctionalization of alkyne derivatives.15–20) In this context, several efficient methodologies for thiocyanation of aromatic compounds have been reported. The major strategy for the purpose is to use the combination of oxidant and inorganic thiocyanate salts, in which either the generation of electrophilic thiocyanium ion equivalent or oxidation of aromatic compounds is the key step.1,21–46)
During the course of our reaction development with hypervalent iodine reagents,47–51) we recently reported several difunctionalizations of alkenes using 1-chloro-1,2-benziodoxol-3-(1H)-one (1).52) Among them, the chlorothiocyanation of alkenes was achieved, in which thiocyanogen chloride (Cl–SCN) was proposed as an active species52) (Chart 1a). Although Cl–SCN is considered to be a highly reactive thiocyanation reagent, there have been only a few reports regarding thiocyanation of aromatic compounds with Cl–SCN.53,54) This is probably because its preparation is technically unfavorable. In general, Cl–SCN can be prepared by using dangerous chlorine gas (Cl2). Another example employed iodobenzene dichloride (Ph–ICl2) and lead(II) thiocyanate (Pb(SCN)2).54,55) Although the reagent is generated under mild conditions, the use of lead compounds should be avoided due to their toxicity. Therefore, we envisioned that our previous method would be an alternative protocol for the reaction with Cl–SCN. In this report, the thiocyanation of aromatic and heteroaromatic compounds using 1 and (trimethylsilyl)isothiocyanate (TMSNCS) is disclosed (Chart 1b).

Chart 1. Thiocyanation Using 1 and TMSNCS
Initially, the reaction conditions were screened with phenol 2a as a test substrate (Table 1). To our delight, TMSNCS was found to be a good thiocyanate source. The reaction in CH2Cl2 occurred at the para-position to give the corresponding product 3a in 92% yield (entry 1). In contrast, low yields were observed in polar solvents (entries 2–4). Potassium thiocyanate could work only in MeOH, albeit in modest yield (entries 5–8). Irrespective of the solvents, thiocyanation product 3a was obtained in modest yields when the reaction was carried out with ammonium thiocyanate (NH4SCN) (entries 9–12). These results suggest that the solubility of the thiocyanate source is important for this thiocyanation.
Table 1. Screening of the Reaction Conditions
a) |
|---|
| Entry | SCN source | Solvent | Yield of 3 [%]b) |
|---|
| 1 | TMSNCS | CH2Cl2 | 92 |
| 2 | TMSNCS | Acetone | 33 |
| 3 | TMSNCS | MeCN | 36 |
| 4 | TMSNCS | MeOH | 6 |
| 5 | KSCN | CH2Cl2 | n.r. |
| 6 | KSCN | Acetone | 4 |
| 7 | KSCN | MeCN | 6 |
| 8 | KSCN | MeOH | 52 |
| 9 | NH4SCN | CH2Cl2 | 64 |
| 10 | NH4SCN | Acetone | 41 |
| 11 | NH4SCN | MeCN | 44 |
| 12 | NH4SCN | MeOH | 48 |
a) The reactions were carried out with 1 (1 equiv.) and thiocyanate source (1 equiv.) on a 0.2 mmol scale at room temperature for 1 h. b) Isolated yield.
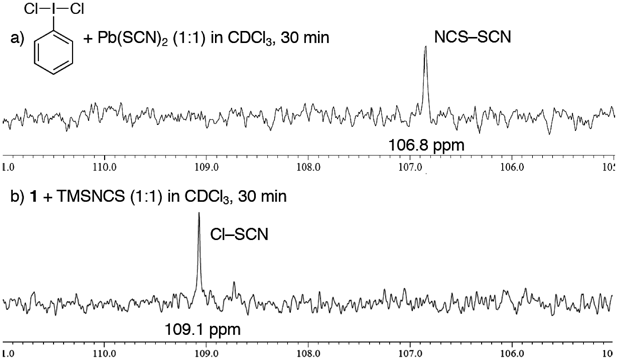
We next tried to confirm the in-situ generated reactive species under the reaction conditions. As seen in our previous report,52) 1H-NMR measurements suggested that Cl–SCN would be generated, because the formation of trimethylsilyl 2-iodobenzoate was observed upon treatment of 1 with 1 equiv. of TMSNCS. In order to obtain more direct evidence, 13C-NMR analysis was carried out56) (Fig. 1). When 1 was reacted with TMSNCS, the peak of TMSNCS disappeared and new peaks were observed at 109.1 ppm (Fig. 1b). The peak of 109.1 ppm was clearly different from thiocyanogen (NCS–SCN, 106.8 ppm), which was prepared according to the literature.26,55) Coupled with the previous 1H-NMR analysis,52) these results strongly indicate that the peak of 109.1 ppm comes from the nitrile carbon of Cl–SCN. After addition of phenol to the mixture, the peak of 109.1 ppm disapperared,56) suggesting Cl–SCN was an active species.
Having established the optimized reaction conditions, the substrate scope was examined (Table 2). Other poly-substituted phenols were good substrates for this reaction and excellent yields were obtained with high para-selectivity (3b–3f). When para-position was substituted by an alkyl group, other positions were thiocyanated. For example, the reaction of 4-tert-butylphenol afforded ortho-thiocyanated product 3g in 69% yield. Interestingly, 3g was relatively unstable even after purification and was gradually converted to cyclized product 3g′.57) Anisole 2h was found to be a good substrate, suggesting that the hydroxyl group is not essential for this reaction. Subsequently, the thiocyanation of aniline derivatives was investigated under the same reaction conditions. As expected, not only anilines but also anilides underwent the reaction smoothly (5a–5d). However, the yield of 5e was only 7%, probably because of the reduced nucleophilicity of 4e. Electron-rich heteroaromatic compounds were also available. Indole gave 3-thiocyanato-1H-indole 7a in 73% yield. The reaction of 2-substituted pyrrole 6b provided 4-thiocyanated product 7b in 61% yield selectively. In contrast to electron-rich aromatic rings, electron-poor aromatic compounds such as quinoline were not reactive at all. Tyrosine derivative 8 was also thiocyanated under the standard conditions. As expected, ortho-substitution of the phenol moiety occurred selectively as in the case of 3g, and the product 9 gradually underwent intramolecular cyclization to 9′.57)
Table 2. Thiocyanation of Various Aromatic and Heteroaromatic Compounds
a)a) The reactions were carried out with 1 (1 equiv.) and TMSNCS (1 equiv.) in CH2Cl2 for 1 h on a 0.2 mmol scale.
To confirm the utility of this protocol, further transformations of the thiocyanation products were demonstrated as shown in Chart 2. Although replacement of the solvent was necessary, the substitution of the cyanide group with a trifluoromethyl group could be carried out in one pot without difficulty.10,36) Another application was [2 + 3] cycloaddition with sodium azide.12) Thus, thiocyanation of 2c followed by [2 + 3] cycloaddition with sodium azide in the presence of ZnCl212) was conducted in one pot, furnishing the desired tetrazole 11 in 37% yield (2 steps).

Chart 2. One-Pot Transformations via Thiocyanation
In summary, we have demonstrated the thiocyanation of aromatic and heteroaromatic compounds using the combination of 1-chloro-1,2-benziodoxol-3-(1H)-one (1) and TMSNCS under mild conditions. Various thiocyanated products were obtained in good to high yields, and the regioselectivity is generally high. The NMR analyses of the mixture of 1 and TMSNCS indicate that thiocyanogen chloride (Cl–SCN) would be the reactive intermediate. In addition, further transformations of the thiocyanated products were successfully demonstrated in one pot. We believe that this protocol would be useful for mild thiocyanation of organic molecules. Further applications are underway in our laboratory.