Regular Articles
Structural Optimization of Caffeoyl Salicylate Scaffold as NO Production Inhibitors
2019 Volume 67 Issue 9 Pages 1006-1014
Details
2019 Volume 67 Issue 9 Pages 1006-1014
Chlorogenic acid (CGA) has been considered as one of important active components in a number of medicinal herbs. Recently our group demonstrated that caffeoyl salicylate scaffold derived from CGA can be employed for the development of novel anti-inflammatory agents. The most active compound D104 can be a very promising starting point for the further structural optimization. A series of novel caffeoyl salicylate analogs were designed, synthesized, and evaluated by preliminary biological evaluation. The obtained results showed that the two compounds B12 and B13 can not only inhibit production of nitric oxide (NO) in RAW264.7 cells induced by lipopolysaccharides (LPS) effectively, but also have high safety in in vitro cytotoxic test, which could be comparable with D104. Molecular docking study on the peroxisome proliferator-activated receptor γ (PPARγ) protein revealed that compounds B12 and B13 can follow the same binding mode with D104, and the carboxyl group of caffeoyl salicylate scaffold might play a key role in the interaction with protein target, which implied the carboxyl group should be retained in the further optimization.
Chlorogenic acid (CGA, Fig. 1) is one of the most available active substances widely distributed in not only lots of Chinese herbal medicine but also in various fruits and vegetables. It has been extensively demonstrated for wide-ranging physiological activities such as anti-obesity,1) anti-diabetic,2) anti-hypertension,3) antimicrobial,4) and anti-inflammatory activities.5–7) Analyzing from the structure, CGA is the simple ester of caffeic acid with quinic acid, two common natural metabolites, which suggests that it has strong potential for chemical modification. CGA could be regarded as a very promising privileged structure for drug discovery.
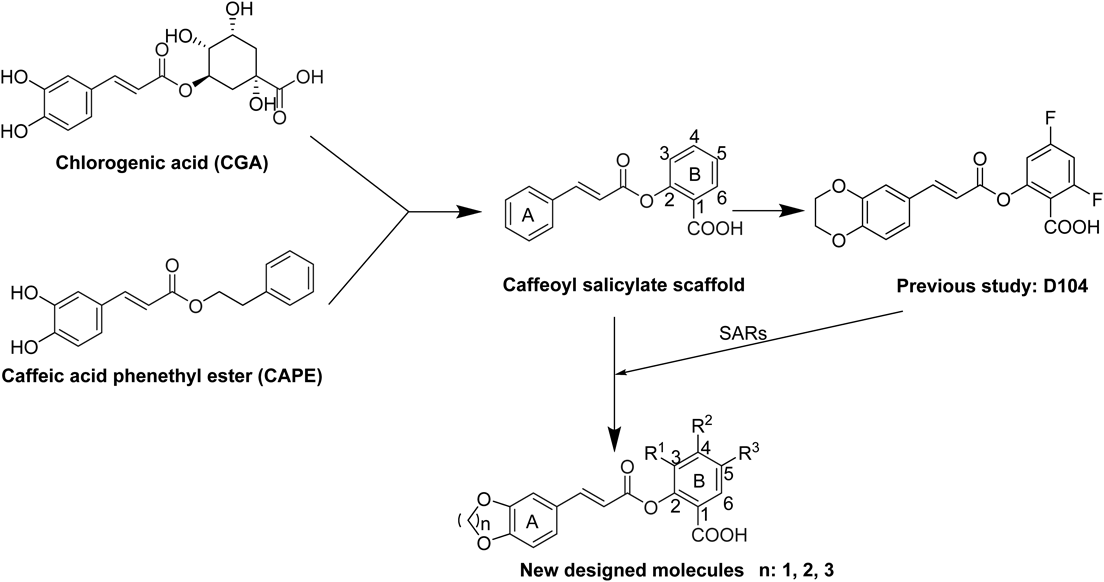
As shown in Fig. 1, we described that the most anti-inflammatory activity compound D104 was screened by a series of caffeoyl salicylate analogs designed by scaffold hopping method based on the chemical structure of CGA and caffeic acid phenethyl ester (CAPE) in the previous study.8) The structure–activity relationships (SARs) of these compounds presents that: A) compounds with 5- or 6-member oxygen-containing ring can exhibit superior potency to compounds with 7- or 8-member oxygen-containing ring; B) electron withdrawing groups of B ring contributed more to anti-inflammatory activity than electron donating groups or no substituent; C) caffeoyl salicylaldehydes generally have potent cytotoxicity, compared to caffeoyl salicylates. Nevertheless, the kinds, quantity and position of substituents seem to be kind of limited. The improvement for anti-inflammatory activity based on the scaffold still has great potential.
In order to further investigate the anti-inflammatory activity of the caffeoyl salicylate scaffold, five different types of substituents including 3,5-, 5-, 3-bromine-5-chlorine, 3-ethoxy, and 4-methoxy, were directly introduced into the B ring of the scaffold. Given the appropriate size of oxygen-containing ring adjacent to the A ring part, the 5-, 6-, and 7-member oxygen-containing ring structures were better to be retained in the new molecules. As presented in Table 1, thirteen novel caffeoyl salicylate analogs in all were designed, synthesized, and evaluated by the corresponding biological assay. Accordingly, the SARs of the scaffold would be further discussed on analysis of these compounds, as well as compounds reported in the previous research.8)
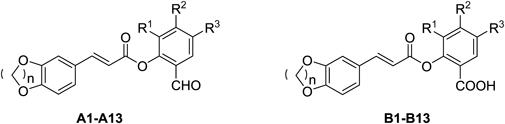 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | n | R1 | R2 | R3 | Cmpd | n | R1 | R2 | R3 |
| A1 | 1 | Br | H | Br | B1 | 1 | Br | H | Br |
| A2 | 2 | Br | H | Br | B2 | 2 | Br | H | Br |
| A3 | 3 | Br | H | Br | B3 | 3 | Br | H | Br |
| A4 | 1 | H | H | Br | B4 | 1 | H | H | Br |
| A5 | 2 | H | H | Br | B5 | 2 | H | H | Br |
| A6 | 3 | H | H | Br | B6 | 3 | H | H | Br |
| A7 | 1 | H | –OCH3 | H | B7 | 1 | H | –OCH3 | H |
| A8 | 2 | H | –OCH3 | H | B8 | 2 | H | –OCH3 | H |
| A9 | 3 | H | –OCH3 | H | B9 | 3 | H | –OCH3 | H |
| A10 | 1 | Br | H | Cl | B10 | 1 | Br | H | Cl |
| A11 | 2 | Br | H | Cl | B11 | 2 | Br | H | Cl |
| A12 | 1 | –OC2H5 | H | H | B12 | 1 | –OC2H5 | H | H |
| A13 | 2 | –OC2H5 | H | H | B13 | 2 | –OC2H5 | H | H |
According to Chart 1, 3,4-dihydroxybenzaldehyde 1 was mixed with the corresponding dibromoalkane and K2CO3 in acetone or N,N-dimethylformamide (DMF) to obtain compounds 2a–2c as the major intermediates. Subsequently, the cyclized cinnamic acids (3a–3c) were prepared by the Knoevenagel condensation reaction, in which compounds 2a–2c can react with malonic acid in the presence of pyridine and piperidine, respectively. After the three cinnamic acids were converted to the corresponding acyl chlorides 4a–4c, the solution of acyl chloride 4a–4c in CH2Cl2 or ethyl acetate (EtOAc) at ice-bath under an inert atmosphere (N2) were added into selected salicylic aldehydes for the synthesis of the required caffeoyl salicylaldehydes. The aldehyde group of compounds A1–A13 can be oxidized to the acid group by the Pinnick oxidation reaction: the caffeoyl salicylates B1–B13 (Table 1) were prepared by the oxidation of compounds A1–A13 under sodium chlorite (NaClO2) in tert-butyl alcohol or tetrahydrofuran (THF). The chemical structures of all new caffeoyl salicylates were characterized by 1H-NMR spectra and general mass spectra (electrospray ionization (ESI)-MS). Besides, all the intermediates such as 2a–2c and 3a–3c had been characterized in the previous study.8)
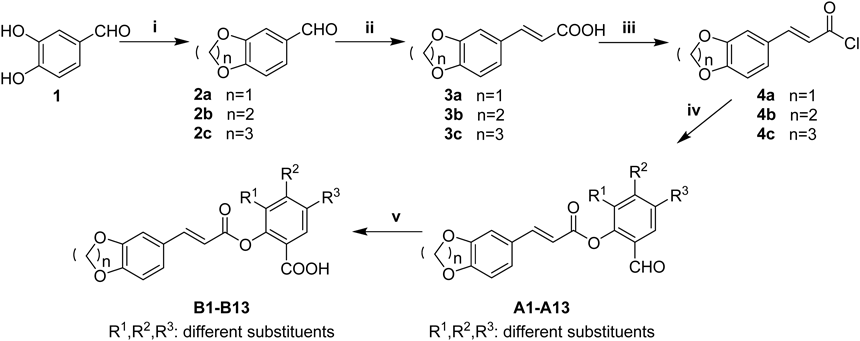
Reagents and conditions: (i) K2CO3, dibromoalkane, acetone or DMF, reflux, 4–5 h; (ii) malonic acid, pyridine, piperidine, 80°C, 24 h; (iii) SOCl2, 80°C, reflux, 2 h; (iv) corresponding salicylic aldehyde, CH2Cl2 or ethyl acetate, pyridine, N2, ice-bath, overnight; (v) NaClO2, NaH2PO4, 3-methyl-1-butene, pH 3–4, stirred at r.t., 5–6 h.
The cytotoxic activities of caffeoyl salicylaldehydes (A1–A13) and caffeoyl salicylates (B1–B13) were evaluated by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method9) using adenocarcinomic human alveolar basal epithelial cells A549 and macrophage cells RAW264.7, respectively. As shown in Fig. 2, most of caffeoyl salicylates exhibited no toxicity against A549 cells when comparing with blank control and two positive molecules (D104 and aspirin). Only compounds B3, B7, and B10 had a bit impact on cell viability, given that the concentration of 50 µM was very high for toxicity test. As expected, the caffeoyl salicylaldehydes (A1–A13) were proved to have potent toxicity against A549 cells, probably due to the high membrane permeability of aldehyde group of these compounds mentioned in the previous study.8) Hence, these caffeoyl salicylates as well as D104 were selected for the further cytotoxicity evaluation against RAW264.7 cells at the corresponding concentration of 25 and 50 µM, respectively. Figure 3 demonstrated most caffeoyl salicylates exhibited no toxicity against RAW264.7 cells, except B7 and B10 that possess low cytotoxicities at high concentration of 50 µM. It is noteworthy that compound B3 has no toxicity against RAW264.7 cells but can inhibit A549 cells, which implied that B3 might have potential antitumor activity.
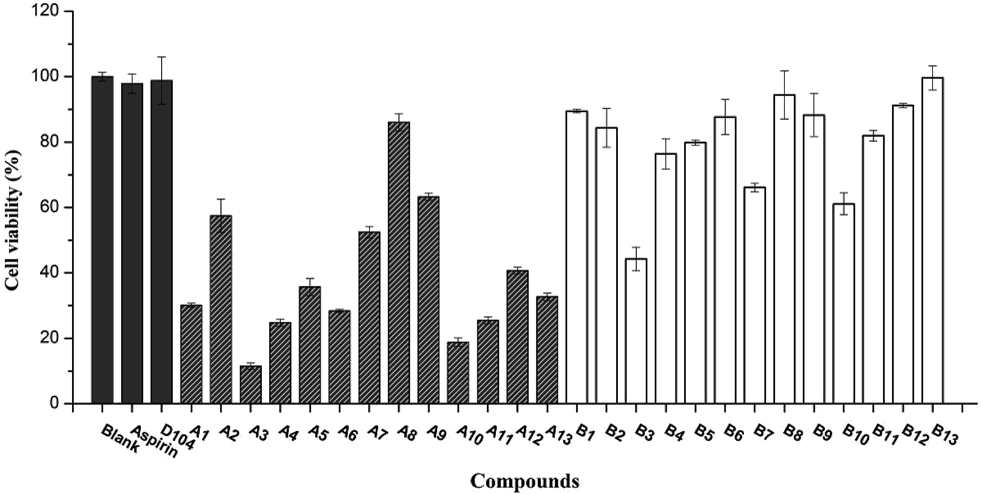
Most caffeoyl salicylaldehyde compounds (A1–A13) did significantly affect cell viability. Data was represented by the mean ± standard error (S.E.) of the three independent experiments.
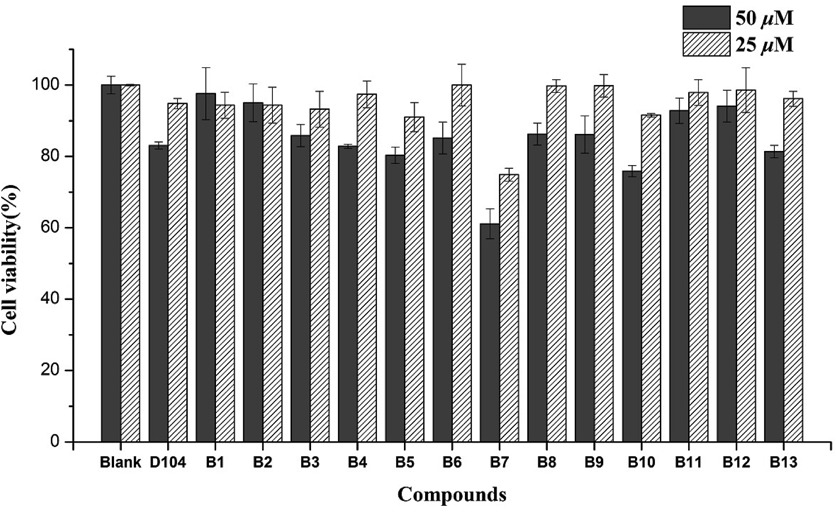
Most caffeoyl salicylate compounds exhibit almost no toxicity except B7 and B10. Data was represented by the mean ± S.D. of the three independent experiments.
Generally, the inhibition of inducible nitric oxide synthase (iNOS) induced by lipopolysaccharide (LPS) can be measured using nitric oxide (NO) release, which to some extent reflects anti-inflammatory activities of these compounds.10) The anti-inflammatory activities of caffeoyl salicylaldehydes (A1–A13) and caffeoyl salicylates (B1–B13) at the concentration of 50 µM can be clearly presented in Fig. 4. In accordance with expectation, all the designed caffeoyl analogs exhibited high-potency inhibitory activity against NO production in LPS-induced RAW264.7 macrophage. However, with regard to caffeoyl salicylaldehydes, the inhibition of NO release should attribute to the high toxicity of caffeoyl salicylaldehydes with high membrane permeability of aldehyde group.
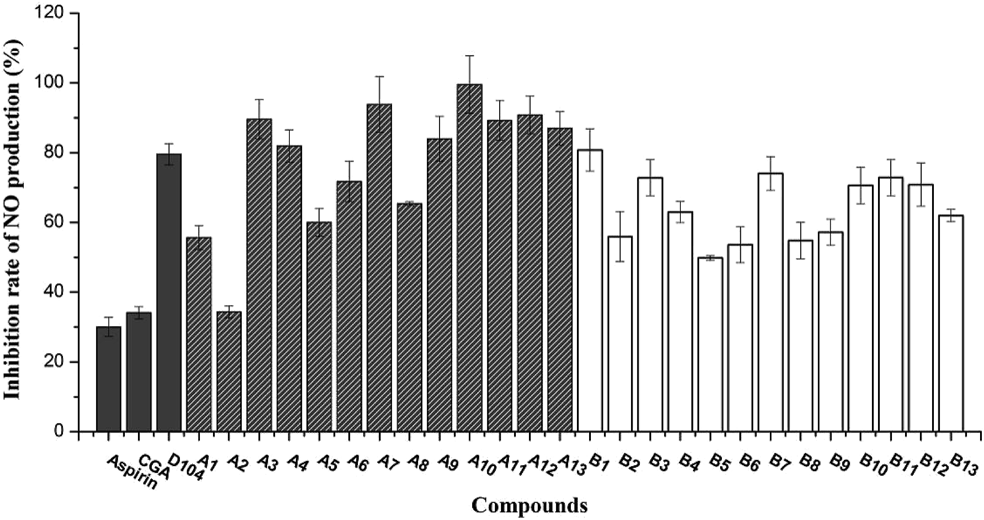
The inhibitory activities of most compounds against NO production were more potent in RAW264.7 when compared to Aspirin and CGA. Data was represented by the mean ± S.D. of the three independent experiments.
As to caffeoyl salicylates (B1–B13), the SARs for inhibition of NO production could be concluded into three points in the previous manuscript: the size of oxygen-containing ring, electron property of substituents, and the number of substituents at the B ring (Fig. 1). Consequently, from analysis of the data presented in Fig. 4, it could be obtained as follows: (1) the size of oxygen-containing ring adjacent to the A ring took obvious effect on inhibitory activity of NO production when comparing compounds B2, B5, B8, B11 and B13 with others; Generally, the inhibition rate of 6-membered-ring derivatives was less than 5- and 7-membered-ring derivatives. (2) compounds with one-substituent of B ring actually displayed more potent inhibitory activity than compounds with double substituents; (3) When introducing one-substituent on B ring and comparing B4–B6 with B7–B9, B12, and B13, we could observe that as follows: a) in terms of substituents positions, introducing groups at the R1 and R2 position exhibited stronger activities than that at the R3 position; b) in consideration of the size of functional groups, bigger substituents (methoxyl and ethyoxyl) might be more beneficial than smaller substituents (bromine and chlorine); c) besides, electron property of substituents could be hardly determined for inhibitory activity of caffeoyl salicylates, and electron withdrawing groups such as chlorine was not necessarily the best.
In addition, ten caffeoyl salicylates except B5, B7, and B10 were investigated for their inhibitory activities of NO production under a decreasing concentration gradient of 100, 50, 25, 12.5, and 6.25 µM. As shown in Fig. 5, most of these compounds can exhibit significant inhibitory activities at high concentrations. Among these compounds, the inhibitory effects of two compounds B12 and B13 against NO production were more potent in RAW264.7 than the positive control D104, even if under low concentrations such as 12.5 or 6.25 µM. Interestingly, the other positive controls Aspirin and CGA can also reach a modest inhibition of NO production at low concentrations, albeit the inhibitory effect under high concentrations seemed to not be obvious. In brief, Fig. 5 presented that all the caffeoyl salicylates can inhibit NO production with a dose-dependent manner, and the two most effective compounds were B12 and B13, respectively, both of which have an ethoxyl at the 3-position of B ring of caffeoyl salicylate scaffold.
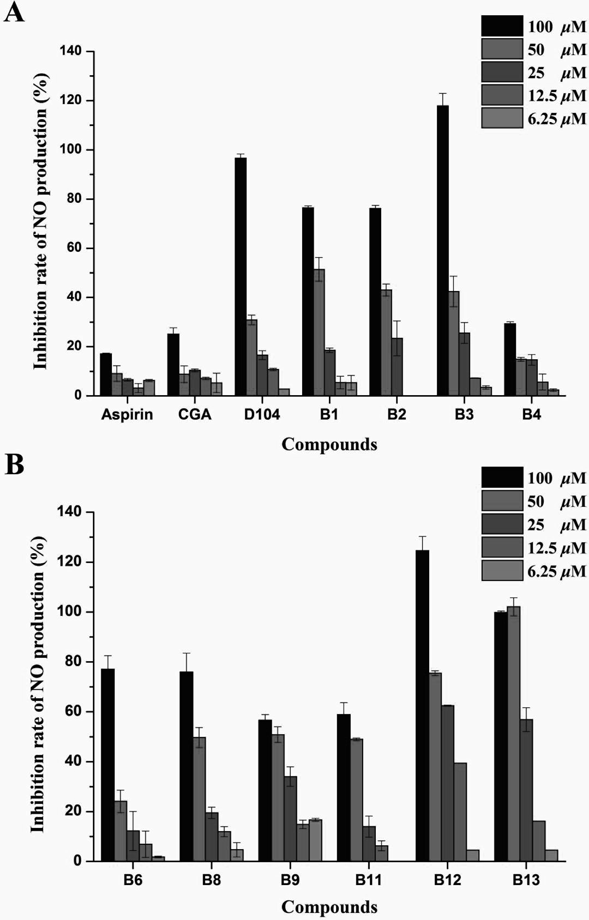
A) Compounds B1–B4 as well as the tree controls above. B) Compounds B6, B8, B9, B11–B13. Data was represented by the mean ± S.D. of the three independent experiments.
Previous study demonstrated that the caffeoyl salicylate scaffold was obtained by virtual screening using the Glide docking study11) on the basis of the peroxisome proliferator-activated receptor γ (PPARγ) protein.12) Accordingly, docking study of the two most active compounds B12 and B13, together with D104 and CGA, were performed to identify the possible binding mode at the active pocket of PPARγ protein (PDB ID: 3U9Q). The docking results can be provided in Fig. 6, which displayed that the three caffeoyl salicylates (D104, B12, and B13) can follow the same binding mode in the pocket, although CGA adopted the reverse orientation completely. Comparing with each other from Fig. 6A to D, the four molecules can insert into the cleft located at the active pocket of PPARγ protein in a linear manner. Besides, the docking scores of B12 and B13 were both better than that of D104.
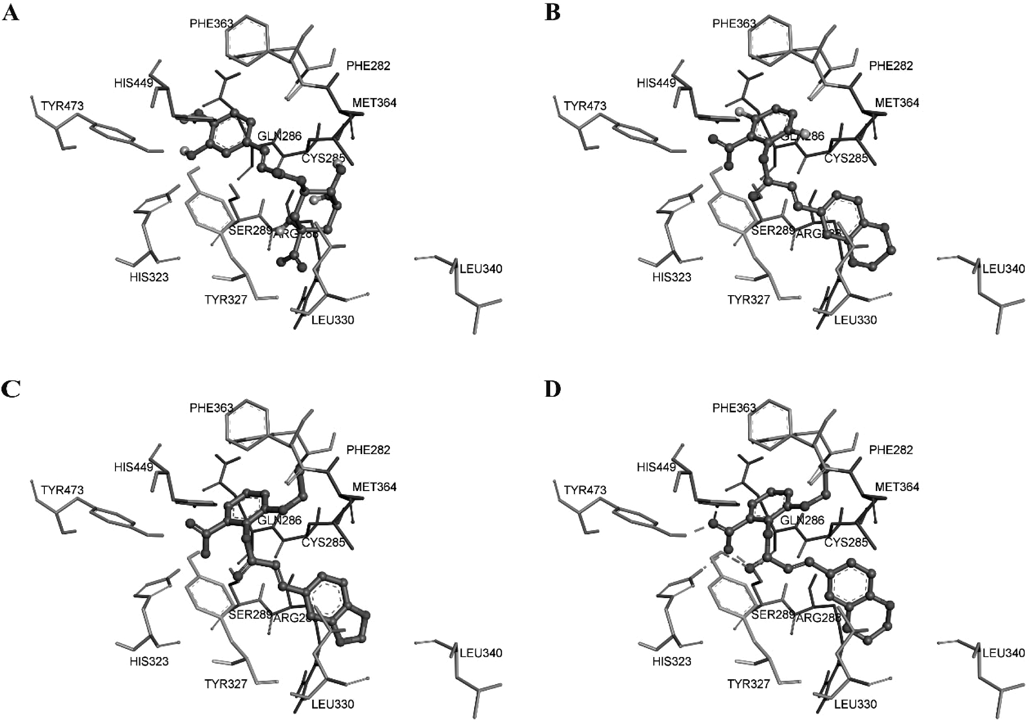
A) Chlorogenic acid (CGA); B) compound D104; C) compound B12; D) compound B13.
More importantly, the local pocket around the B ring of caffeoyl salicylates seemed enough for providing the further optimization space of this scaffold. This is the reason why compounds with various substituents still exhibited potent inhibitory activity against NO production, unless the volume of substituent was too large such as the N,N-diethylamine group of compounds 6d, 6h, and 6l in the previous paper,8) which may lead to the decrease of inhibitory activity. In addition, the carboxyl group of B ring can form three hydrogen bonds with HIS323, TYR473 of the binding site, and ester group of compounds itself, respectively. Therefore, we can infer that the carboxyl group of caffeoyl salicylate scaffold would play a key role in the interaction with protein target, and should be retained in the further optimization.
Recently our group demonstrated that caffeoyl salicylate scaffold derived from CGA can be regarded as one promising starting template for the development of novel anti-inflammatory agents. Accordingly, on the basis of the primary SARs of the scaffold, a series of novel caffeoyl salicylate analogs were designed, synthesized, and evaluated by preliminary biological evaluation. The obtained results showed that the two compounds B12 and B13 can not only inhibit production of NO in RAW264.7 cells induced by LPS effectively, but also have high safety in in vitro cytotoxic test, which can be better than D104. Molecular docking study on the PPARγ protein revealed that compounds B12 and B13 can follow the same binding mode with D104, and the carboxyl group of caffeoyl salicylate scaffold might play a key role in the interaction with protein target, which implied the carboxyl group should be retained in the further optimization.
1H spectra were recorded on Bruker AM 600 MHz spectrometers with tetramethylsilane (TMS) as the internal standard. ESI-MS and high-resolution (HR)-MS were recorded by Agilent 6520B Q-TOF. Melting points (mp) were recorded on SRS OptiMelt-100 full automatic micro melting point instrument. Analytical HPLC was performed on a Agilent 1260 HPLC system using a Chromolith SpeedROD RP-18 column (4.6 ×50 mm). A linear gradient elution was performed with eluent A (H2O/TFA, 100 : 0.01) containing 0% of solvent B (CH3CN/H2O/TFA, 90 : 10 : 0.01) rising to 100% of B during 20 min with a flow rate of 1.0 mL/min. Column chromatography (CC): silica gel (200–300 mesh; Qingdao Makall Group Co., Ltd.; Qingdao; China). All reactions were monitored using TLC on silica gel plates. Reaction reagents were analytical reagent grade and purchased from Aladdin.
General Procedures for the Synthesis of Compounds 2a–2cAll the procedures and spectral data of compounds 2a–2c can refer to the previous paper.8)
General Procedures for the Synthesis of Compounds 3a–3cAll the procedures and spectral data of compounds 3a–3c can refer to the previous paper.8)
General Procedures for the Synthesis of Compounds 4a–4cAll the procedures of compounds 4a–4c can refer to the previous paper.8)
General Procedures for the Synthesis of Compounds A1–A13A solution of acyl chloride (12 mM) in CH2Cl2 or ethyl acetate was added dropwise to corresponding salicylic aldehyde (10 mM) in CH2Cl2 or ethyl acetate containing pyridine (1.6 mL, 20 mM) under an inert (N2) atmosphere and at 0°C with constant stirring overnight. The reaction mixture was then poured in excess of diluted NaOH and extracted with CH2Cl2 or ethyl acetate. The extraction liquid was purified by a flash chromatography with ethyl acetate/petroleum ether to give these compounds. The yields were between 40% and 60%.
2,4-Dibromo-6-formylphenyl(E)-3-(benzo[d][1,3]dioxol-5-yl)acrylate (A1)Mp 240–242°C. MS (ESI): 450.89 (C17H10Br2O5, [M−H]−). 1H-NMR (600 MHz, dimethyl sulfoxide-d6 (DMSO-d6)) δ: 9.95 (s, 1H), 8.39 (d, J = 2.4 Hz, 1H), 8.12 (dd, J = 3.5, 2.4 Hz, 2H), 7.92 (d, J = 2.5 Hz, 1H), 7.88 (d, J = 15.9 Hz, 1H), 7.57 (d, J = 1.7 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.85 (d, J = 15.9 Hz, 1H), 6.12 (s, 2H). Purity by anal. HPLC: 96% (254 nm).
2,4-Dibromo-6-formylphenyl(E)-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylate (A2)Mp 248–250°C. MS (ESI): 464.91 (C18H12Br2O5, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 9.95 (s, 1H), 8.39 (d, J = 2.4 Hz, 1H), 8.12 (d, J = 2.4 Hz, 1H), 7.85 (d, J = 15.9 Hz, 1H), 7.43 (d, J = 2.1 Hz, 1H), 7.35 (dd, J = 8.4, 2.1 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.82 (d, J = 15.9 Hz, 1H), 4.32–4.30 (m, 2H), 4.30–4.27 (m, 2H). Purity by anal. HPLC: 95% (254 nm).
2,4-Dibromo-6-formylphenyl(E)-3-(3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)acrylate (A3)Mp 256–258°C. MS (ESI): 478.92 (C19H14Br2O5, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 9.95 (s, 1H), 8.38 (d, J = 2.4 Hz, 1H), 8.12 (d, J = 2.4 Hz, 1H), 7.87 (d, J = 16.0 Hz, 1H), 7.49 (d, J = 2.2 Hz, 1H), 7.45 (dd, J = 8.4, 2.2 Hz, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.85 (d, J = 16.0 Hz, 1H), 4.22 (t, J = 5.6 Hz, 2H), 4.18 (t, J = 5.6 Hz, 2H), 2.14 (p, J = 5.6 Hz, 2H). Purity by anal. HPLC: 95% (254 nm).
4-Bromo-2-formylphenyl(E)-3-(benzo[d][1,3]dioxol-5-yl)acrylate (A4)Mp 182–184°C. MS (ESI): 372.98 (C17H11BrO5, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 10.03 (s, 1H), 8.07 (d, J = 2.5 Hz, 1H), 7.96 (dd, J = 8.6, 2.6 Hz, 1H), 7.83 (d, J = 15.9 Hz, 1H), 7.54 (d, J = 1.7 Hz, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.32 (dd, J = 8.1, 1.7 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 15.9 Hz, 1H), 6.12 (s, 2H). Purity by anal. HPLC: 97% (254 nm).
4-Bromo-2-formylphenyl(E)-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylate (A5)Mp 190–192°C. MS (ESI): 386.99 (C18H13BrO5, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 10.03 (s, 1H), 8.07 (d, J = 2.5 Hz, 1H), 7.96 (dd, J = 8.6, 2.6 Hz, 1H), 7.80 (d, J = 15.9 Hz, 1H), 7.41–7.38 (m, 2H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.77 (d, J = 16.0 Hz, 1H), 4.31 (dd, J = 5.5, 2.2 Hz, 2H), 4.28 (dd, J = 5.5, 2.2 Hz, 2H). Purity by anal. HPLC: 97% (254 nm).
4-Bromo-2-formylphenyl(E)-3-(3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)acrylate (A6)Mp 199–201°C. MS (ESI): 401.01 (C19H15BrO5, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 10.03 (s, 1H), 8.07 (d, J = 2.6 Hz, 1H), 7.96 (dd, J = 8.6, 2.6 Hz, 1H), 7.82 (d, J = 16.0 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.42 (dd, J = 8.3, 2.2 Hz, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.80 (d, J = 16.0 Hz, 1H), 4.21 (t, J = 5.6 Hz, 2H), 4.18 (t, J = 5.6 Hz, 2H), 2.14 (p, J = 5.6 Hz, 2H). Purity by anal. HPLC: 97% (254 nm).
2-Formyl-5-methoxyphenyl(E)-3-(benzo[d][1,3]dioxol-5-yl)acrylate (A7)Mp 156–158°C. MS (ESI): 325.08 (C18H14O6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 9.92 (s, 1H), 7.88 (d, J = 8.7 Hz, 1H), 7.81 (d, J = 15.9 Hz, 1H), 7.53 (d, J = 1.7 Hz, 1H), 7.31 (dd, J = 8.0, 1.7 Hz, 1H), 7.06 (dd, J = 8.7, 2.4 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 2.4 Hz, 1H), 6.79 (d, J = 15.9 Hz, 1H), 3.88 (s, 3H), 3.29 (s, 2H). Purity by anal. HPLC: 95% (254 nm).
2-Formyl-5-methoxyphenyl(E)-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylate (A8)Mp 164–166°C. MS (ESI): 339.09 (C19H16O6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 9.92 (s, 1H), 7.87 (d, J = 8.6 Hz, 1H), 7.78 (d, J = 15.9 Hz, 1H), 7.38 (s, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.05 (d, J = 8.6 Hz, 1H), 6.98 (m, 2H), 6.75 (d, J = 16.0 Hz, 1H),4.30 (t, 2H), 4.28 (t, J = 5.4 Hz, 2H), 3.88 (s, 3H). Purity by anal. HPLC: 95% (254 nm).
2-Formyl-5-methoxyphenyl(E)-3-(3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)acrylate (A9)Mp 171–173°C. MS (ESI): 353.11 (C20H18O6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 9.92 (s, 1H), 7.87 (d, J = 8.7 Hz, 1H), 7.80 (d, J = 16.0 Hz, 1H), 7.45 (d, J = 2.1 Hz, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.06 (dd, J = 8.7, 2.4 Hz, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.96 (d, J = 2.4 Hz, 1H), 6.78 (d, J = 16.0 Hz, 1H), 4.23–4.20 (m, 2H), 4.19 (t, J = 5.6 Hz, 2H), 3.88 (s, 1H), 2.15 (t, J = 5.6 Hz, 2H). Purity by anal. HPLC: 97% (254 nm).
2-Bromo-4-chloro-6-formylphenyl(E)-3-(benzo[d][1,3]dioxol-5-yl)acrylate (A10)Mp 211–213°C. MS (ESI): 406.94 (C17H10BrClO5, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 9.97 (s, 1H), 8.28 (d, J = 2.5 Hz, 1H), 8.00 (d, J = 2.6 Hz, 1H), 7.88 (d, J = 15.9 Hz, 1H), 7.56 (d, J = 1.7 Hz, 1H), 7.34 (dd, J = 8.1, 1.7 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.84 (d, J = 15.9 Hz, 1H), 6.12 (s, 2H). Purity by anal. HPLC: 95% (254 nm).
2-Bromo-4-chloro-6-formylphenyl(E)-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylate (A11)Mp 218–220°C. MS (ESI): 420.96 (C18H12BrClO5, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 9.97 (s, 1H), 8.28 (d, J = 2.6 Hz, 1H), 8.00 (d, J = 2.6 Hz, 1H), 7.85 (d, J = 16.0 Hz, 1H), 7.42 (d, J = 2.1 Hz, 1H), 7.35 (dd, J = 8.4, 2.1 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.81 (d, J = 15.9 Hz, 1H), 4.32 (t, 2H), 4.29 (t, 2H). Purity by anal. HPLC: 95% (254 nm).
General Procedures for the Synthesis of Compounds B1–B13A mixture of A1–A13 (0.15 g, 0.5 mmol) and tert-butyl alcohol (8 mL) and THF (6 mL) was stirred at room temperature until clear, and 3-methyl-1-butene (0.84 mL, 10 mM) that was cooled at 0°C was added. Sodium dihydrogen phosphate (0.6 g, 5 mM) and NaClO2 was dissolved in 2 mL aqueous solution and acidified with hydrochloric acid aqueous solution (3 mM) to pH 3–4. The solution was slowly added into the prepared aldehyde. Then the reaction mixture was stirred at room temperature for 5–6 h. The solvent was evaporated away and purified by a flash chromatography in pleasing yield (Yield: 45–60%).
(E)-2-((3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)oxy)-3,5-dibromobenzoic Acid (B1)Mp 226–228°C. MS (ESI): 466.88 (C17H10Br2O6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.27 (d, J = 2.4 Hz, 1H), 8.03 (d, J = 2.4 Hz, 1H), 7.78 (d, J = 15.9 Hz, 1H), 7.41 (d, J = 2.1 Hz, 1H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.78 (d, J = 15.9 Hz, 1H), 4.30 (t, J = 3.7 Hz, 2H). Purity by anal. HPLC: 97% (254 nm).
(E)-3,5-Dibromo-2-((3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acryloyl)oxy)benzoic Acid (B2)Mp 234–236°C. MS (ESI): 480.90 (C18H12Br2O6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.25 (d, J = 2.5 Hz, 1H), 8.03 (d, J = 2.4 Hz, 1H), 7.78 (d, J = 16.0 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 15.9 Hz, 1H), 4.34 –4.30 (m, 2H), 4.28–4.26 (m, 2H). Purity by anal. HPLC: 97% (254 nm).
(E)-3,5-Dibromo-2-((3-(3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)acryloyl)oxy)benzoic Acid (B3)Mp 242–244°C. MS (ESI): 494.92 (C19H14Br2O6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.58 (s, 1H), 8.38 (s, 1H), 8.15 (d, J = 16.0 Hz, 1H), 7.82 (s, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.15 (d, J = 16.0 Hz, 1H), 4.57 (t, J = 5.5 Hz, 2H), 4.54 (t, J = 5.6 Hz, 2H), 2.86 (s, 2H). Purity by anal. HPLC: 96% (254 nm).
(E)-2-((3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)oxy)-5-bromobenzoic Acid (B4)Mp 220–222°C. MS (ESI): 388.97 (C17H11BrO6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.02 (d, J = 2.6 Hz, 1H), 7.85 (dd, J = 8.5, 2.6 Hz, 1H), 7.75 (d, J = 15.9 Hz, 1H), 7.53 (d, J = 1.7 Hz, 1H), 7.29 (dd, J = 8.1, 1.7 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.76 (d, J = 15.9 Hz, 1H), 6.11 (s, 2H). Purity by anal. HPLC: 96% (254 nm).
(E)-5-Bromo-2-((3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acryloyl)oxy)benzoic Acid (B5)Mp 227–229°C. MS (ESI): 402.99 (C18H13BrO6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.00 (s, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.71 (d, J = 15.9 Hz, 1H), 7.35 (s, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.69 (d, J = 15.9 Hz, 1H), 4.29 (d, J = 13.9 Hz, 4H). Purity by anal. HPLC: 95% (254 nm).
(E)-5-Bromo-2-((3-(3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)acryloyl)oxy)benzoic Acid (B6)Mp 235–237°C. MS (ESI): 417.01 (C19H15BrO6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.38 (s, 1H), 8.21 (d, J = 7.8 Hz, 1H), 8.10 (d, J = 16.0 Hz, 1H), 7.79 (s, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.62 (d, J = 8.6 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.10 (d, J = 16.0 Hz, 1H), 4.56 (t, J = 6.0 Hz, 2H), 4.54 (t, J = 5.3 Hz, 2H), 2.86 (s, 2H). Purity by anal. HPLC: 95% (254 nm).
(E)-2-((3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)oxy)-4-methoxybenzoic Acid (B7)Mp 193–195°C. MS (ESI): 341.07 (C18H14O7, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 7.89 (d, J = 8.6 Hz, 1H), 7.72 (d, J = 15.9 Hz, 1H), 7.53 (d, J = 1.7 Hz, 1H), 7.51 (d, J = 1.7 Hz, 1H), 7.31 (dd, J = 8.1, 1.7 Hz, 2H), 7.06 (dd, J = 8.7, 2.5 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 2.5 Hz, 1H), 6.11 (s, 3H), 3.88 (s, 4H). Purity by anal. HPLC: 97% (254 nm).
(E)-2-((3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)acryloyl)oxy)-4-methoxybenzoic Acid (B8)Mp 201–203°C. MS (ESI): 355.09 (C19H16O7, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 7.91 (d, J = 8.8 Hz, 1H), 7.69 (d, J = 16.0 Hz, 1H), 7.36 (d, J = 2.1 Hz, 1H), 7.29 (dd, J = 8.4, 2.1 Hz, 1H), 6.96–6.90 (m, 2H), 6.83 (d, J = 2.6 Hz, 1H), 6.70 (d, J = 16.0 Hz, 1H), 4.31–4.29 (m, 2H), 4.29–4.26 (m, 2H), 3.84 (s, 3H). Purity by anal. HPLC: 97% (254 nm).
(E)-2-((3-(3,4-Dihydro-2H-benzo[b][1,4]dioxepin-7-yl)acryloyl)oxy)-4-methoxybenzoic Acid (B9)Mp 209–211°C. MS (ESI): 369.11 (C20H18O7, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 12.48 (s, 1H), 7.91 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 16.0 Hz, 1H), 7.43 (d, J = 2.2 Hz, 1H), 7.39 (dd, J = 8.3, 2.2 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.95 (dd, J = 8.8, 2.6 Hz, 1H), 6.83 (d, J = 2.5 Hz, 1H), 6.73 (d, J = 16.0 Hz, 1H), 4.19 (dt, J = 15.0, 5.5 Hz, 4H), 3.84 (s, 3H), 2.17–2.10 (m, 2H). Purity by anal. HPLC: 97% (254 nm).
(E)-2-((3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)oxy)-3-bromo-5-chlorobenzoic Acid (B10)Mp 196–198°C. MS (ESI): 422.93 (C17H10BrClO6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.15 (s, 1H), 7.91 (d, J = 2.6 Hz, 1H), 7.82 (d, J = 15.8 Hz, 1H), 7.54 (s, 1H), 7.32 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 15.9 Hz, 1H), 6.11 (s, 2H). Purity by anal. HPLC: 96% (254 nm).
(E)-3-Bromo-5-chloro-2-((3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acryloyl)oxy)benzoic Acid (B11)Mp 204–206°C. MS (ESI): 436.95 (C18H12BrClO6, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 8.15 (s, 1H), 7.91 (d, J = 2.6 Hz, 1H), 7.78 (d, J = 16.0 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 15.9 Hz, 1H), 4.31–4.30 (m, 2H), 4.28–4.27 (m, 2H). Purity by anal. HPLC: 96% (254 nm).
(E)-2-((3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)oxy)-3-ethoxybenzoic Acid (B12)Mp 205–207°C. MS (ESI): 355.09 (C19H16O7, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 12.88 (s, 1H), 7.73 (dd, J = 15.9, 2.0 Hz, 1H), 7.52 (d, J = 1.7 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.35 (dd, J = 8.3, 1.7 Hz, 1H), 7.31 (d, J = 7.9 Hz, 1H), 7.28 (dd, J = 7.9, 1.8 Hz, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.76 (dd, J = 15.9, 1.5 Hz, 1H), 6.10 (s, 2H), 4.07 (q, J = 7.0 Hz, 2H), 1.26 (t, J = 7.0 Hz, 3H). Purity by anal. HPLC: 97% (254 nm).
(E)-2-((3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)acryloyl)oxy)-3-ethoxybenzoic Acid (B13)Mp 212–214°C. MS (ESI): 369.11 (C20H18O7, [M−H]−). 1H-NMR (600 MHz, DMSO-d6) δ: 12.90 (s, 1H), 7.70 (d, J = 16.0 Hz, 1H), 7.44 (dd, J = 7.7, 1.6 Hz, 1H), 7.37 (d, J = 2.1 Hz, 1H), 7.35 (d, J = 6.7 Hz, 1H), 7.32–7.29 (m, 2H), 6.92 (d, J = 8.3 Hz, 1H), 6.73 (d, J = 16.0 Hz, 1H), 4.30–4.29 (m, 2H), 4.28–4.27 (m, 2H), 1.25 (t, J = 6.9 Hz, 3H). Purity by anal. HPLC: 97% (254 nm).
RAW264.7 cells and A549 cells were obtained from State Key Laboratory of Medicine, Nanjing University. LPS, MTT, and Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, and 2% phosphoric acid) were purchased from Sigma. The 6- and 96-well plates were purchased from Beyotime Biotechnology.
RAW264.7 cells and A549 cells were grown in High glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin and propagated at 37°C in a humidified atmosphere containing 5% CO2 in air.
Compounds A1–A13, B1–B13, D104, CGA, and acetylsalicylic acid (Aspirin) were dissolved in dimethyl sulfoxide to make stock solutions, respectively, and kept at −20°C. The final concentration of the vehicle in the solution never exceeded 0.1% and had no effects on NO production and cell viability.
Assay for Cytotoxic ActivityCompounds A1–A13, B1–B13, and D104 were assayed according to the MTT test9) under the concentration of 50 µM against A549 cells. Moreover, compounds B1–B13 and D104 were also chosen to test the cytotoxic effects against RAW264.7 cells under the two concentrations (25 and 50 µM). MTT was dissolved at 4 mg/mL in phosphate buffered saline (PBS) and used essentially as previously described. Briefly, cell lines in logarithmic phase were seeded at a density of 3 × 103 cells/well in 100 µL of DMEM into 96-well microtiter plates. After 24 h, exponentially growing cells were exposed to the indicated compounds at various concentrations. After 48 h in final volumes of 200 µL, cell survival was determined by the addition of an MTT solution (20 µL of 4 mg/mL MTT in PBS) for 4 h. After carefully removing the medium, the precipitates were dissolved in 200 µL of DMSO, shaken mechanically for 10 min, and then absorbance values at a wavelength of 540 nm were taken on a SpectraMax 190 microplate reader (Molecular Devices, U.S.A.). Survival ratios are expressed in percentages with respect to untreated cells.
Anti-inflammatory AssayAccumulation of nitrite (NO2−), an indicator of NO synthase activity, in culture supernatant fluids was measured based on Griess reaction.13) Briefly, Cells (2 × 104) were seeded in 100 µL of DMEM into 96-well plates and co-incubated with different concentrations (6.25, 12.5, 25, 50, 100 µM) of compounds B1–B4, B6, B8, B9, B11–B13, D104, CGA, and Aspirin in the absence or presence of LPS (500 ng/mL) for 48 h. Meanwhile, chlorogenic acid and acetylsalicylic acid also were tested as positive controls. Culture supernatant fluids were mixed with 100 µL Griess reagent at room temperature for 5 min. Using NaNO2 to generate a standard curve, nitrite production was measured by an absorbance reading at 540 nm.
Compounds B12, B13, D104, and CGA were imported into the LigPreb module of the Schrodinger 2015 suite. The PPARγ protein crystal complex (PDB ID: 3U9Q) had been downloaded from the PDB website,14) and prepared in Protein Preparation Wizard. Subsequently, four prepared small molecules, together with PPARγ protein, were imported into the GLIDE module integrated in the Schrodinger 2015 suite. The docking study was performed at the GLIDE standard precision (SP) mode. All the docking parameter was set default values.
This work was supported by the Natural Science Foundation of Jiangsu Higher Education Institutions (Grant number: 18KJB350004), the Project for Young Teachers of Nanjing Forestry University (Grant number: CX2017005), and the Doctorate Fellowship Foundation of Nanjing Forestry University (Grant number: 163030748).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.