Regular Articles
Novel Triazole Derivatives Containing Different Ester Skeleton: Design, Synthesis, Biological Evaluation and Molecular Docking
2020 年 68 巻 1 号 p. 64-69

詳細
2020 年 68 巻 1 号 p. 64-69
Invasive fungal disease constitutes a growing health problem and development of novel antifungal drugs with high potency and selectivity are in an urgent need. In this study, a novel series of triazole derivatives containing different ester skeleton were designed and synthesized. Microdilution broth method was used to investigate antifungal activity. Significant inhibitory activity of compounds 5c, 5d, 5e, 5f, 5m and 5n was evaluated against the Candida albicans (I), Candida albicans clinical isolate (II), Candida glabrata clinical isolate (I), and Candida glabrata (II) with minimum inhibitory concentrations (MIC80) values ranging from 2 to 16 µg/mL. Notably, compounds 5e and 5n showed the best inhibition against Candida albicans (II), Candida glabrata (I), and Candida glabrata (II) at the concentrations of 2 and 8 µg/mL, respectively. Molecular docking study revealed that the target compounds interacted with CYP51 mainly through hydrophobic and van der Waals interactions. The results indicated that these novel triazole derivatives could serve as promising leads for development of antifungal agents.
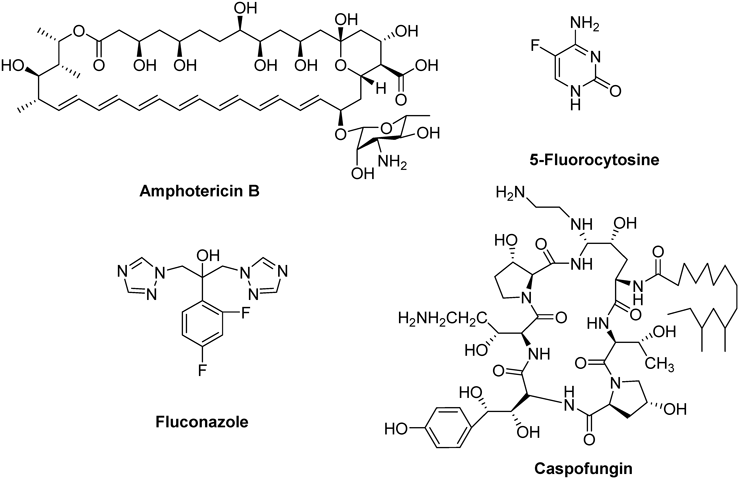
Global morbidity due to life-threatening fungal infections has dramatically increased in recent decades, particularly among individuals suffering from cancer, autoimmune diseases, organ transplants or AIDS.1–3) Clinically, Aspergillus fumigatus (mortality rate: 50–90%), Cryptococcus neoformans (mortality rate: 20–70%), and Candida albicans (mortality rate: 20–40%) are the major pathogenic agents for systemic fungal infections.4) Numerous antifungal drugs including polyenes (e.g., amphotericin B), fluorinated pyrimidines (e.g., 5-fluorocytosine), azoles (e.g., fluconazole), and echinocandins (e.g., caspofungin) (Fig. 1) are available for the treatment of invasive fungal infections.5) For their high therapeutic index, broad spectrum activities and superior safety profile, the azole antifungals are considered as first-line therapy in current clinic. However, some of them still suffer from serious drug resistance issues, there is a real need for developing novel antifungal triazoles with improved profiles.6,7)
Azole antifungal agents competitively inhibit the lanosterol 14α-demethylase (CYP51), which catalyzes the oxidative removal of the 14α-methyl group of lanosterol to give △14,15-desaturated intermediates in ergosterol biosynthesis. This selective inhibition of CYP51 would result in depletion of ergosterol and accumulation of lanosterol, thereby inhibiting the growth and proliferation of fungi.8,9) Moreover, CYP51 has been regarded by many researchers as a target for molecular docking and widely applied in the rational design of antifungal compounds.10–15)
Studies showed that16) the antifungal triazoles featured a core structure composed of a triazole, a halophenyl ring, while differing in the side chains. The development of triazole drugs is primarily driven through structure optimization especially of the side chains as well as structure–activity relationship (SAR) study. Itraconazole (Fig. 2) is the triazole antifungal drug available in the market and widely used for candidiasis. The long side chain of itraconnazole containing four linearly linked cyclic rings might attribute to its low solubility and erratic bioavailability, which drove the research to design novel agents with medium side chains.17) Therefore, replacing the long side chain in the itraconazole structure with different ester moieties containing aryl rings and halogenated alkyl chain might result in novel triazole antifungal drugs. Furthermore, molecular docking study indicated that the triazole bound to the heme iron of CYP51, and the halophenyl ring could locate into the hydrophobic pocket.18) We hypothesized that the ester moieties might accommodate in the deep cavity of CYP51, as well as providing in hydrophobic pockets of the biological targets which contribute to improve the potency, stability and specificity of the binding site. In addition, the ester groups were to be introduced in the target compounds because they could be further removed to reduce molecular weight. The halogen as a strong electron-withdrawing group could affect the charge distribution and hydrophobility of drug molecules and the time of drug action. Thus, the halogenated alkyl chain was to be introduced to leave large space for further optimization.

The above mentioned promoted us to design and synthesize a series of 1-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethyl derivatives containing a triazole ring, a dichlorophenyl group, and ester groups in the side chain. In our design, the structure of itraconazole was altered as a platform and different ester groups were insert into the side chain to find out whether these groups were necessary for the antifungal activity (Fig. 2).
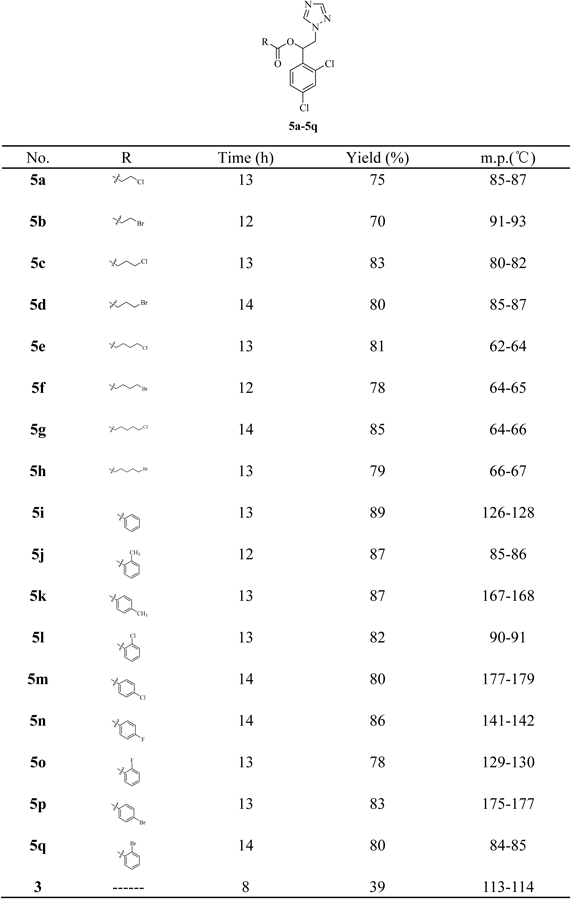
1-(2,4-Dichlorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethyl derivatives (5a–5q) were synthesized in four steps as illustrated in Chart 1. At first, 1,3-dichlorobenzene (1.0 equiv), chloroacetyl chloride (ClCH2COCl) (1.2 equiv) and anhydrous aluminum chloride (AlCl3) (1.0 equiv) in an anhydrous dichloromethane (DCM) solution were heated at reflux for 4 h to obtain compound 2 in 80% yield.19) Secondly, intermediate 3 was prepared by condensation of compound 2 (1.0 equiv) with triazole (1.5 equiv) in the presence of potassium carbonate (K2CO3) (1.0 equiv) and acetonitrile (CH3CN) for 8 h at reflux. Intermediates 3 was obtained in the yield of 39%. Thirdly, intermediate 3 (1.0 equiv) and sodium borohydride (NaBH4) (2.0 equiv) in an anhydrous ethanol–tetrahydrofuran (THF) solution were reacted at room temperature for 4 h to obtain compound 4 in 80% yield. Finally, compound 4 (1.0 equiv) and various acid (1.5 equiv) were reacted in the presence of N,N′-dicyclohexylcarbodiimide (DCC) (1.3 equiv), 4-dimethylaminopyridine (DMAP) (1.3 equiv) and anhydrous dichloromethane for overnight at reflux to give final compounds 5a–5q in 70–89% yield. The reaction was monitored by TLC, and the reaction mixture was purified by column chromatography.

Reagents and conditions: (i) ClCH2COCl, AlCl3, anhydrous CH2Cl2, reflux, 4 h, 80% yield; (ii) Triazole, K2CO3, CH3CN, reflux, 8 h, 39% yield; (iii) NaBH4, Ethanol–THF, r.t., 4 h, 80% yield; (iiii) DCC, DMAP, anhydrous CH2Cl2, reflux, overnight, 70–89% yield.
The structures of all compounds were shown in Table 1. All the compounds were elucidated by 1H-NMR, 13C-NMR and high resolution (HR) MS.
 |
The antifungal activity of the compounds 5a–5q against nine human pathogenic fungi was evaluated according to the protocols of the National Committee for Clinical Laboratory Standards. Fluconazole (FCZ), Amphotericin B (Amp B) and Itraconazole (ITZ) were used as positive control drugs. The broth microdilution method was used to determine the minimum inhibitory concentrations (MIC80) of the synthesized compounds.20)
As shown in Table 2, most of the compounds showed weak antifungal activity against C. par., A. fum., C. kru. and C. tro., while there was a good antifungal activity against C. alb., C. gla. and C. neo. Several compounds, such as compounds 5c, 5d, 5e, 5f, 5m and 5n exhibited superior or comparable antifungal activity to FCZ with MIC80 values in the range of 2–16 µg/mL, particularly for C. alb. (I), C. alb. (II), C. gla. (I), and C. gla. (II). Among them, compounds 5e and 5n showed the most potent activity against four important fungal pathogens (MIC80 = 2–8 µg/mL). Compound 5c (inhibition of C. alb. (II)) could be achieved at 2 µg/mL and compounds 5d and 5f (inhibition of C. alb. (II)) could be achieved at 4 µg/mL. Compound 5m (inhibition of C. gla. (I) and C. gla. (II)) could be achieved at 8 µg/mL. These above-mentioned compounds exhibited superior activity to FCZ. Interestingly, compound 5p (MIC80 = 2 µg/mL) was highly active against C. alb. (I), which was more active than FCZ (MIC80 = 8 µg/mL).
| Compd. | MIC80 (µg/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C. alb. (I) | C. alb. (II) | C. kru. | C. par. | C. gla. (I) | C. gla. (II) | C. neo. | A. fum. | C. tro. | |
| 5a | 64 | 16 | 128 | 64 | 128 | 128 | 128 | 64 | >128 |
| 5b | 64 | 16 | 128 | 64 | 128 | 128 | 128 | 128 | >128 |
| 5c | 8 | 2 | 64 | 32 | 16 | 16 | 32 | 128 | >128 |
| 5d | 8 | 4 | 128 | 64 | 16 | 16 | 32 | 64 | >128 |
| 5e | 4 | 2 | 128 | 128 | 8 | 8 | 32 | 128 | >128 |
| 5f | 8 | 4 | 128 | 128 | 16 | 16 | 32 | 128 | >128 |
| 5g | 32 | 16 | >128 | 64 | 32 | 32 | 32 | 128 | >128 |
| 5h | 64 | 16 | >128 | 64 | 64 | 32 | 64 | 128 | >128 |
| 5i | 64 | 32 | >128 | 128 | 128 | 128 | 128 | 128 | >128 |
| 5j | 128 | 64 | >128 | 128 | >128 | 128 | 128 | 128 | >128 |
| 5k | 16 | 32 | >128 | 128 | 32 | 128 | 128 | 128 | >128 |
| 5l | 128 | 64 | >128 | 128 | 128 | 128 | 128 | 64 | >128 |
| 5m | 8 | 8 | >128 | 64 | 8 | 8 | 128 | 128 | >128 |
| 5n | 2 | 2 | 32 | 64 | 8 | 8 | 128 | 128 | >128 |
| 5o | 128 | 64 | 128 | 128 | 128 | 128 | 128 | 128 | >128 |
| 5p | 2 | 32 | 128 | >128 | 16 | 32 | 128 | 128 | >128 |
| 5q | 128 | 64 | 64 | >128 | 128 | 128 | 128 | 128 | >128 |
| 3 | 32 | 32 | >128 | 64 | 64 | 64 | >128 | 128 | >128 |
| 4 | 16 | 32 | 128 | 16 | 128 | 128 | 64 | 64 | >128 |
| FCZ | 8 | 8 | 64 | 16 | 16 | 16 | 32 | 128 | 32 |
| ITZ | 0.25 | 0.25 | 1 | 0.5 | 2 | 2 | 8 | 4 | 1 |
| Amp B | <1 | <1 | <1 | <1 | <1 | 2 | 1 | 2 | <1 |
a) Abbreviations: C. alb. (I), Candida albicans (ATCC 90028); C. alb. (II), Candida albicans clinical isolate (1802274); C. kru., Candida krusei (ATCC 6528); C. par., Candida parapsilosis clinical isolate (18022019); C. gla. (I), Candida glabrata clinical isolate (1802183); C. gla. (II), Candida glabrata (ATCC 15126); C. neo., Cryptococcus neoformans (ATCC 34877); A. fum., Aspergillus fumigatus clinical isolate (2423); C. tro., Candida tropicalis clinical isolate (1802372); FCZ: Fluconazole; ITZ: Itraconazole; Amp B: Amphotericin B.
In contrast, the target compounds generally showed improved activity against C. neo., whereas they were less potent against C. par. For example, the inhibitory activity of compounds 5c, 5d, 5e, 5f and 5g (MIC80 = 32 µg/mL) against C. neo. were comparable to that of FCZ (MIC80 = 32 µg/mL). For C. kru., most compounds showed weak activity except compound 5n (MIC80 = 32 µg/mL), which was more potent than FCZ (MIC80 = 64 µg/mL). However, all the compounds were inactive against A. fum. and C. tro.
As seen from the Table 2, compound 3 and intermediate 4 showed weak antifungal activity as compared to the compounds 5a–5q. The introduction of different ester groups on the intermediate 4 resulted in a better improvement of the antifungal activity.
SARs StudyOn the basis of the antifungal activity, preliminary SARs were obtained. Firstly, compared with the compounds 3 and 4, the introduction of different acids on the 1-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethan-1-one scaffold (5a–5q) resulted in a better improvement of the antifungal activity, suggesting that the interactions of the aliphatic and aromatic groups with CYP51 were important for its binding affinity.
Secondly, for the compounds 5a–5h, the length of halogenated alkyl side chain on ester group was essential to their antifungal activities. The data suggested that the optimal chain lengths for maximal antifungal activity were C4 and C5 (5c, 5d, 5e, 5f), and the general trend for activity versus chain length was C5 > C4 > C6 > C3. Among the compounds 5c, 5d, 5e, 5f, chloro substituted alkyl side chain on ester group (5c, 5e) was more active than the bromo substituted (5d, 5f) against C. alb., C. gla. and C. neo.
Thirdly, for the compounds 5i–5q, electron-withdrawing groups (halogens, such as -F, -Cl and -Br) on benzene ring, enhanced the inhibitory activity against C. alb., C. gla. and C. neo. On the contrary, electron-donating groups (such as –CH3), decreased the inhibitory activity. Therefore, to some extent, we could predict that electron-withdrawing group could affect the charge distribution and hydrophobility of drug molecules to improve the biological activity of compounds. Moreover, 4-substitutions on benzene ring (5m, 5n and 5p) were more favorable than the 3-substitutions (5l, 5o and 5q). Among the compounds 5m, 5n, 5p, 4-fluoro substitution (5n) was more active than the 4-chloro substitution (5m). However, 4-bromo substitution (5p) had little effect on the antifungal activity as compared to the compounds 5n and 5m. These results gave us a clear conclusion that the halogen substituents on benzene ring gave rise to different antifungal activity depending on the halogen position and type.
Finally, the replacement of the halogenated alkyl ester side chains (5a–5h) by phenyl groups (5i–5q) resulted in a decrease in antifungal activity. These results clearly indicated the importance that halogen substituents have on the antifungal activity of this class of triazole derivatives.
Molecular DockingIn order to rationalize the antifungal activity and provide more straightforward information about ligand-enzyme interactions for further rational drug design, molecular docking was performed for itraconazole (ITZ), compounds 5e and 5q. The crystal structure of CYP5121) (PDB ID: 5EQB) bound with its active ligand ITZ was served as a useful template for generating proposed binding modes. The docking results were showed in Fig. 3 and Table 3.
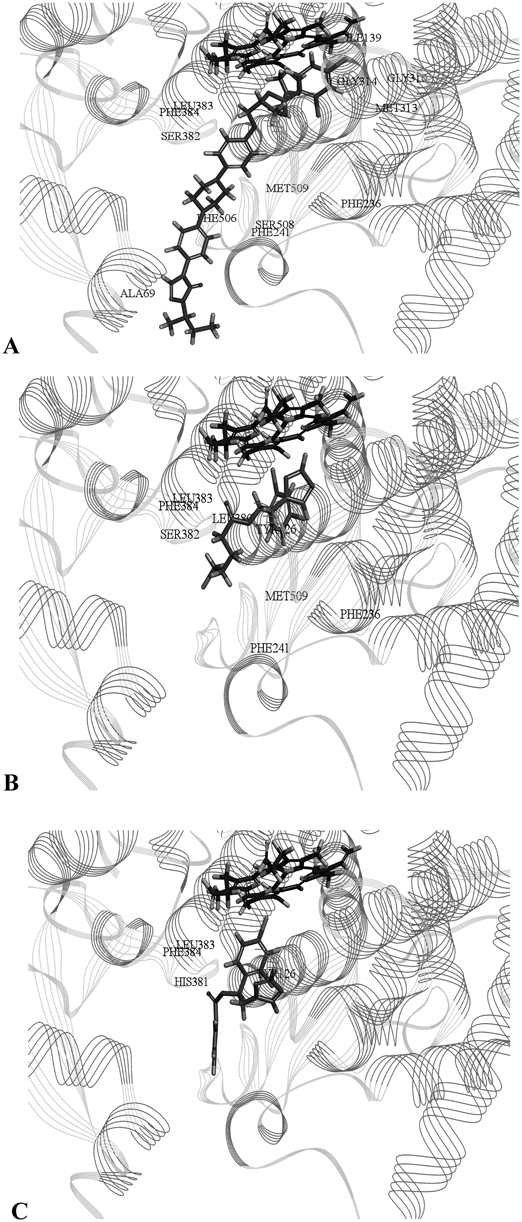
The heme is shown in dark gray (upper) and molecules ITZ, 5e and 5q are shown in gray (lower).
| LIGANDS | –CDOCKER ENERGYa) (kcal/mol) | –CDOCKER INTERACTION ENERGYb) (kcal/mol) |
|---|---|---|
| Itraconazole | 14.2919 | 80.1265 |
| 5h | 27.1352 | 48.3584 |
| 5e | 21.4974 | 45.1817 |
| 5f | 21.6036 | 44.9928 |
| 5g | 25.7246 | 43.1719 |
| 5c | 23.5840 | 40.9958 |
| 5d | 19.3272 | 40.8749 |
| 5m | 15.9591 | 40.5997 |
| 5p | 9.7196 | 40.5280 |
| 5a | 18.0549 | 40.0338 |
| 5l | 4.0308 | 39.9109 |
| 5k | 17.2615 | 39.7354 |
| 5j | 11.9170 | 39.5278 |
| 5b | 20.2553 | 39.4440 |
| 5n | 14.1954 | 38.6478 |
| 5q | 3.6624 | 38.1424 |
| 5o | 11.6228 | 38.0478 |
| 5i | 12.2871 | 37.5572 |
| 3 | 6.8256 | 32.2895 |
a) –CDOCKER ENERGY: The higher the value of –CDOCKER ENERGY, the more stable the conformation. b) –CDOCKER INTERACTION ENERGY: The higher the CDOCKER INTERACTION ENERGY value represents more stable ligand binding.
The overall conformation of the ITZ in the active site of CYP51 was shown in Fig. 3A. The docking method was quite accurate to predict the real interaction of ligands and CYP51, where the re-docking experiment gave a RMSD value of 1.9844 Å. Moreover, the triazole group of the ITZ made a coordination bond with the heme iron and showed carbon hydrogen bond interaction with Gly314. The 2,4-dichlorophenyl group could be put into the hydrophobic pocket formed by Ile139, Met313 and Phe236. In addition, the long tail of ITZ lied within a hydrophobic pocket and formed van der Waals interactions with the surrounding residues, such as Leu383, Ser382, Ala69 and Phe384. The phenyl group of side chain could form π-π interaction with Phe241 and the piperazine ring of side chain had carbon hydrogen bonds with residues of Ser508 and Met509.
More insights revealed that compound 5e might bind with the active site cavity of CYP51 (Fig. 3B). The –CDOCKER ENERGY value of it, which was 21.4974 kcal/mol, was higher than the natural ligand ITZ (14.2919 kcal/mol). It indicated that compound 5e might exert better antifungal activity than ITZ. The triazole ring made a coordination bond with the heme iron and formed hydrogen bond interaction with Tyr126. The 2,4-dichlorophenyl group could show π–π interactions with Leu380 and Phe236. Further, the ester side chain was interacting with Met509, Leu383, Phe384, Phe241 and Ser382 through van der Waals force.
Compared with compound 5e, compound 5q showed weaker antifungal activity. The calculated –CDOCKER ENERGY value was 3.6624 kcal/mol. The –CDOCKER INTERACTION ENERGY value of it, which was 38.1424 kcal/mol, was lower than the compound 5e (45.1817 kcal/mol). As shown in Fig. 3C, the triazole ring showed π–π interaction with His381 and the ester side chain was interacting with Phe384, Tyr126 and Leu383 through Van der Waals force. Compound 5q showed weaker interactions with the key amino acid residues of the CYP51 binding pocket than compound 5e. Thus, the molecular modeling studies suggested that compound 5e might act as an antifungal agent by occupying the CYP51 binding pocket and showing favorable interactions with the key amino acid residues.
A novel series of triazole derivatives containing different ester skeleton were synthesized and evaluated as antifungal agents. Compounds 5e and 5n showed the best antifungal activity in vitro, which (inhibition of Candida albicans (II) and Candida glabrata (I), (II)) could be achieved at the concentrations of 2 and 8 µg/mL, respectively. In addition, compounds 5c, 5d, 5f and 5m displayed the remarkable in vitro inhibitory activity against Candida albicans and Candida glabrata strains, which was superior or comparable to that of the reference drug fluconazole. However, all the compounds also showed weak inhibition to other fungi which were Candida parapsilosis, Aspergillus fumigatus, Candida krusei and Candida tropicalis. Moreover, molecular docking was performed to position compounds 5e and 5q into the CYP51 active site to determine the potential binding mode. In conclusion, these preliminary results are promising and some of these compounds may be potential candidates as antifungal agents.
General chemistry methods, synthesis procedures, spectral data, biological assays, molecular docking are given in supplementary materials.
We gratefully acknowledge The Independent Scientific Research Project Program of Ordos Central Hospital (EY2017002) for financial support; The Joint Project of Science and Technology Million Engineering of Inner Mongolia Medical University (YKD2017KJBW(LH)023); The Natural Science Foundation of Hebei Province (B2018201269).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.