Abstract
Novel reactions using hetero-heavy atoms (P, S, Si, Se, and Sn) were developed and applied to the synthesis of biofunctional molecules and some medicine-candidates, in which the following items are covered. 1) Development of introduction of C1-unit using cyanophosphates (CPs). 2) Carbene-generation under neutral condition from CPs and its application to organic synthesis. 3) [3,3]Sigmatropic rearrangement-ring expansion reactions of medium-sized cyclic thionocarbonates containing a sulfur atom and their application to natural product synthesis. 4) Stereoselective synthesis of novel β-imidazole C-nucleosides via diazafulvene intermediates and their application to investigating ribozyme reaction mechanism. 5) Developments of novel histamine H3- and H4-receptor ligands using new synthetic methods involving Se or Sn atoms.
1. Introduction
Organic synthesis has reached a high degree of perfection. However, reactions occurring within a living organism are much more mild, and more highly efficient, with specificity and selectivity. Thus, new efficient synthetic methods and reagents should be developed further. Although catalysts now occupy a central position in organic synthesis, reagents remain important, and many novel and improved reagents have been developed. The exploitation of useful reagents and methods can often make the impossible possible, and it is the starting point for the development of new fields in organic synthesis as well as their practical and industrial applications.
Diethyl phosphorocyanidate [(EtO)2P(O)CN, DEPC] was developed by Shioiri in 1972 and is a useful and versatile reagent for organic synthesis,1) in particular as a condensing reagent for the synthesis of amides and peptides (Chart 1), and for its application in the total synthesis of macrolactams which are present in various marine natural products.1)

Chart 1. Synthesis of Amides and Peptides Using DEPC
The present author first encountered DEPC, which contains a phosphorus atom (a hetero-heavy atom), as a graduate student at Prof. Shioiri’s laboratory of Nagoya City University. Since the DEPC structure contains a CN, the role of DEPC as a cyanating reagent under non-aqueous conditions could be expected. The author started to investigate novel cyanations using DEPC under his supervision, and it was my first job as a chemist. The study on cyanation using DEPC revealed the following non-aqueous reactions, that is, reaction of enamines with DEPC to afford α-amino nitriles, the function of which is easily converted into α-amino acids2) (Chart 2, Eq. 1); modified Strecker synthesis for the preparation of α-amino nitriles using DEPC from carbonyl compounds and amines in tetrahydrofuran (THF) under mild reaction conditions (Eq. 2)3); modified Reissert–Henze reaction, in which DEPC reacts with the aromatic amine oxide in the presence of triethylamine to give the α-cyanated product (Eq. 3)4); reaction of sodium sulfinates with DEPC to give thiocyanates, providing a novel route to thiocyanate synthesis (Eq. 4).5–7) These reactions contrast to conventional cyanation procedures employing alkaline cyanides (NaCN/KCN) in aqueous solvents.

Chart 2. Non-aqueous Cyanation Using DEPC
Through these studies, we addressed new reactions using hetero-heavy atoms (P, S, Si, Se, and Sn) having vacant d-orbital contributions. More importantly, the developed reactions were applied to the efficient synthesis of natural products, biofunctional molecules and some medicine-candidates. This review describes important aspects in the author’s work over the last three decades, in which the following studies are covered: 1) C1-unit introduction via cyanophosphates as well as carbene-generation from cyanophosphates under neutral condition and their application to organic synthesis. 2) [3,3]Sigmatropic rearrangement-ring expansion reaction of medium-sized cyclic thionocarbonates and application to synthesis of natural products. 3) Stereoselective synthesis of β-imidazole C-nucleosides and its application to investigating ribozyme reaction mechanism. 4) Development of novel histamine H3 and H4 receptor ligands using new reactions characterized by Se or Sn atoms.
2. C1-Unit Introduction Using Cyanophosphates
2.1. Formation of Cyanophospates from Carbonyl Compounds Using DEPCCyanohydrins 1 and O-trimethylsilyl (TMS)-protected derivatives 2 are well-known versatile synthetic intermediates in organic synthesis8) (Fig. 1). To date, TMS-cyanohydrins 2 have been demonstrated useful precursors for the preparation of carbonyl anion synthons, cyanohydrins, β-amino alcohols, and other functional compounds.8,9) However, the intrinsic instabilities of cyanohydrins and TMS derivatives have limited their potential application. We therefore investigated the formation of cyanohydrin-O-phosphates (cyanophosphates: CPs) 3 as novel protected cyanohydrins, which were expected to be diverse cyanation intermediates due to their high functional density and the electrophilic character of the phosphate leaving group. Examination of the literature revealed that CPs had been previously obtained by treating cyanohydrins with dialkyl phosphorochloridate, and some CPs were of particular use as agricultural chemicals, for example, as insecticides, fungicides, nematocides, and the like.10)
In 1983, we first observed a cyanophosphorylation reaction on treatment of ketones or aldehydes with DEPC in the presence of a catalytic amount of lithium diisopropylamide (LDA) in THF11) (Chart 3). Although 1 eq. of DEPC was generally employed, 3 eq. DEPC was beneficial for sluggish reactions. Various ketones and aldehydes (4 and 5, respectively) react with DEPC to give CPs 3 in good yields.

Chart 3. Cyanophosphorylation in the Presence of A Catalytic Amount of LDA
A proposed mechanism for the cyanophosphorylation of ketones in the presence of LDA suggested that the treatment of ketones with DEPC in the presence of a catalytic amount of LiCN would produce CPs.1,10) This working hypothesis therefore led to a convenient synthetic procedure for the formation of CPs 3 using DEPC (3 eq.) and LiCN (3 eq.) as outlined in Chart 4.12)

Chart 4. Cyanophosphorylation of Ketones 4 or Aldehydes 5 Using DEPC (3 eq.) and LiCN (3 eq.)
The LiCN method afforded CPs 3 from a range of ketones 4 and aldehydes 5 in excellent yields at room temperature (r.t.) under neutral conditions (c.f., the basic conditions required for the LDA method).11) Indeed, this method has now been adopted in the majority of CP synthetic pathways.1) Furthermore, excess DEPC (3 eq.) and LiCN (3 eq.) are now used routinely for cyanophosphorylation,12) although we have more recently demonstrated that the use of 1.2 eq. DEPC and 0.6 eq. LiCN is sufficient for the formation of CP from ketones, while 1.2 eq. DEPC and 0.1 eq. LiCN are required when aldehydes are employed, as shown in Table 1.13)
Table 1. Cyanophosphorylation of Carbonyl Compounds Using DEPC (1.2 eq.) and LiCN (Cat)
Since our early reports on the cyanophosphorylation of carbonyl compounds,11,12) many synthetic procedures of CPs were reported by use of modified reagent systems; diethyl phosphorochloridate (DEPCl)-LiCN (Nájera et al.), DEPCl-NaCN (Shin et al.), DEPC-Et3N in the solvent-free system (Nájera et al.), and DEPC-N-heterocyclic carbene (NHC) catalyst (Kondo et al. and Dai et al).1) However, the DEPC-LiCN method has been used as the simplest method for CPs in most cases.1) Furthermore, two groups of Nájera and Shibasaki reported independently the enantioselective cyanophosphorylation of aldehydes.1)
2.2. Synthesis of α,β-Unsaturated NitrilesThe majority of cyclic alkenenitriles are synthesized from cyclic ketone precursors via conversion to the corresponding cyanohydrins or their derivatives followed by dehydration.1) Due to the diethyl phosphate anion being a good leaving group, benzylic CPs 7 prepared from aromatic ketones 6 were readily converted into α,β-unsaturated nitriles 8 by treatment with boron trifluoride etherate (BF3·OEt2) in benzene at rt in high yields (Table 2), in which the transformation revealed an efficient synthetic route to α,β-unsaturated nitriles from aromatic ketones.12) 4-Isobutylacetophenone (6e) afforded α-cyanostyrene 8e (80%) (entry 5 in Table 2). As an application of this method, 8e was subsequently subjected to hydrogenation followed by hydrolysis to give (±)-ibuprofen, an anti-inflammatory agent, in 70% overall yield from 6e.12) In the case of diketone 6f bearing both aromatic and aliphatic ketones, the CP function derived from the aliphatic ketone remained intact under BF3·OEt2 treatment, giving 8f in 92% yield (entry 6).12)
Table 2. Synthesis of α,β-Unsaturated Nitriles from Ketones
via Benzylic CPs
Kurihara et al. carried out cyanophosphorylation on Uhle’s ketone (9) using DEPC and LiCN followed by treatment with BF3·OEt2 to afford α,β-unsaturated nitrile 11 (90%).14,15) Compound 11 was then reduced using diisobutylaluminium hydride (DIBAL) to give the aldehyde 12, which was successfully transformed into (±)-lysergic acid (13) over 4 steps, as outlined in Chart 5.14,15)

Chart 5. Synthesis of (±)-Lysergic Acid from Uhle’s Ketone
Furthermore, in the synthetic applications of the cyanophosphorylation–dephosphorylation method, this method provided crucial unsaturated nitriles, which led to many bioactive compounds: A-80426 (an antidepressant combining α-2 antagonism with serotonin (5-HT) uptake inhibition); (±)-lunarine (a macrocyclic polyamine alkaloid); a dicyanoflubene dimer as an electron-accepting molecule; L-histidine with high 13C incorporation; spiropiperidines using the formation of α,β-unsaturated nitrile by kinetic dephosphorylation.1)
2.3. Reductive Cyanation of Ketones and AldehydesThe transformation of carbonyl groups into nitriles represents a useful one-carbon homologation for the synthesis of a variety of carboxylic acid and amine derivatives. As one of the strategies, CPs have been employed as intermediates to achieve this transformation. For example, CPs 3 can be hydrogenated over Pd/C, treated with lithium and ammonia, or reduced mildly with samarium iodide (SmI2) to give homologous nitriles 141) (Chart 6).

Chart 6. Reductive Cyanation of Carbonyl Compounds via CPs
Synthesis of arylacetonitriles (R1 = Ar) via hydrogenolysis over Pd/C of arylketone- or arylaldehyde-CPs in ethyl acetate was efficiently applied for the synthesis of a range of non-steroidal anti-inflammatory agents, including ibuprofen, naproxen, and clidanac.16,17) Particularly, in the preparation of loxoprofen (18), diketone 15 was easily converted to bis-CP 16, which was selectively dephosphorylated under standard hydrogenolysis conditions to give mono-CP 17, which was then hydrolyzed to give loxoprofen (18).16,17) In this case, the CP functional group in the cyclopentanone moiety played a significant role in protection of the carbonyl group (Chart 7).

Chart 7. Synthesis of Loxoprofen
In addition, we investigated dephosphorylation of CPs by reduction with lithium in liquid ammonia, as outlined in Table 3.18,19) Reduction of a CP derived from citral 19 with lithium in liquid ammonia at −78°C and quenching of the reaction mixture with isoprene afforded β,γ-unsaturated nitrile 20 (86%, entry 1, Table 3), while quenching of the CP derived from α-ionone (21) using octyl iodide yielded the α-alkylated nitrile 22 (83%, entry 2). Furthermore, reduction of the CP derived from 4-isobutylacetophenone (23) under refluxing conditions (at −33°C) followed by quenching with ammonium chloride (NH4Cl) yielded 1-ethyl-4-isobutylbenzene (24, 89%)(entry 3), thus giving a deoxygenated derivative of an aromatic ketone.19) Interestingly, no isomerized or reduced alkenes were formed in the reduction stages.18,19) Thus, these reductive dephosphorylation reactions have potential for application in the preparation of CPs derived from enones and aromatic ketones.1)
Table 3. Reduction of CPs
3 with Lithium in Liquid Ammonia
We further developed the reductive cyanation of CPs using SmI2, which is a moderate reducing agent20,21) (Table 4). This method also yielded nitriles chemoselectively from various carbonyl compounds possessing multiple and sensitive functionalities via CPs (entries 1–6, Table 4). For example, cyanation of epoxy aldehyde 25 was conducted by a combination of cyanophosphorylation and SmI2 reduction followed by recyclization of the oxirane ring-opened product with triethylamine, to give the epoxy nitrile 26 in 54% yield (entry 7).20) This reaction was successfully applied to α,β-unsaturated carbonyl compounds under neutral conditions to obtain the desired nitriles. Cholest-4-en-3-one (27) and α-ionone (29) gave the corresponding nitriles 28 and 30 (entries 9 and 10, Table 4), whereas under general tosylmethyl isocyanide (TosMIC)22) conditions in the presence of t-BuOK in DMF, nitriles 28 and 30 were detected only by TLC in trace amounts.20)
Table 4. Reductive Cyanation of CPs with SmI
2a) The product was prepared by TosMIC method.
The CPs-SmI2 method was employed in a variety of organic syntheses,1) for instance, in the synthesis of the natural spongian pentacyclic diterpene, aplyroseol-1 and its related compounds; in the synthesis of hydroxyethene isosteres of human immunodeficiency virus (HIV) protease-catalyzed phenylalanine (Phe)-proline (Pro) hydrolysis transition state; in the transformation of aldol product from Boc-L-phenylalaninal into lactone; in the synthesis of sweroside aglycon; and in the preparation of nitriles from chiral ketone in the total synthesis of (±)-aphidicolin.
2.4. Allylic Rearrangement of CPsThe CPs (B) derived from α,β-unsaturated ketones (A) were transformed into conjugated allylic phosphates (C) via BF3·Et2O-catalyzed allylic rearrangement23,24) (Chart 8). In the case of 2-cyclohexen-1-one (31), cyanophosphorylation followed by treatment of the resulting CP 32 with a catalytic amount of BF3·OEt2 (0.1 eq.) afforded 3-cyanocyclohex-2-enyl diethyl phosphate (33) in 81% yield23,24) (Chart 8).

Chart 8. Allylic Rearrangement of CPs to Give Allylic Phosphates
From a stereochemical viewpoint, 6-methylbicyclo[4,4,0]dec-1-en-3-one (34) can be converted into CP 35 bearing a 3α-cyano-3β-diethylphosphooxy group. Subsequent treatment of 35 with BF3·OEt2 provided a diene nitrile 37 (90%), as shown in Chart 9.23) Alternatively, heating CP 35 at reflux in benzene in the absence of BF3·Et2O produced 38 (46%), bearing a 5β-diethylphosphooxy group, along with the accompanying diene nitrile 37 (50%). Phosphate 38 could be converted into 37 in quantitative yield by treatment with BF3·OEt2. In addition, the formation of both 35 and 38 indicated that the rearrangement took place via a [3,3]-sigmatropic rearrangement in a suprafacial fashion, as shown at 36 in Chart 9.23,24)

Chart 9. Stereochemistry of Allylic Rearrangement of CPs
Two decades after our initial report,23) Shibasaki and co-workers achieved efficient asymmetric synthesis of oseltamivir phosphate (Tamiflu),25) an important anti-influenza drug, via allylic rearrangement of a CP, as illustrated in Chart 10.25)

Chart 10. Asymmetric Synthesis of Oseltamivir Phosphate Based on Allylic Rearrangement of CP
In continuation of our recent program on the use of CPs, we were encouraged to look once again at allylic rearrangements of CPs for a practicable synthetic method for key intermediate 41 for 3-(tetrazol-5-yl)-3,5-pregnadien-20-one (TzPD, 43), a potent 5α-reductase inhibitor (IC50: 15.6 nM), from pregnene-3,20-dione (39) in 92% overall yield in four steps, as shown in Chart 1126) Furthermore, it should be noted that the C20-CP of bis-CP 40 also plays a significant role as a protecting group for the C20-ketone.

Chart 11. Synthesis of TzPD 43 from Pregnene-3,20-dione (39)
3. Tetrazole-Fragmentation for Generation of Alkylidene Carbenes from CPs under Neutral Conditions and Its Application to Organic Synthesis27)
3.1. Overview of Alkylidene Carbene-GenerationAlkylidene carbenes (44) can be used in many synthetically valuable reactions including [1,2]-migration, ylide formation, alkyne cyclopropanation and [1,5]-C–H bond insertion.28,29) Notably, [1,2]-migrations occurring with alkylidene carbenes (44) are useful for preparation of homologous alkynes (45), which are extensively used in organic synthesis28,29) (Chart 12, Eq. 1). Meanwhile, alkylidene carbenes (46) having a long alkyl chain undergo regioselective and stereoselective [1,5]-C–H bond insertions, which are well suited for the synthesis of cyclopentenes (47, X = CH2) (Eq. 2).28,29) In the case that a heteroatom is present in the connecting chain, five-membered unsaturated heterocycles (47, X = O or N-R) are produced. The high reactivity of alkylidene carbenes necessitates in situ generation, typically via the use of diazo olefination reagents, haloalkenes, or alkynyliodonium salts.28,29) One of the most widely employed methods to access alkylidene carbenes is loss of N2 from a double bond terminus, particularly from 1-diazoalkenes. These reactions employ dimethyl(diazomethyl)phosphonate esters [(MeO)2P(O)CHN2: DAMP] (Seyferth–Gilbert reagent)30–32) or trimethylsilyldiazomethane (TMSCHN2)33) (Chart 13, Eq. 1), but require strongly basic conditions, which is a potential drawback. Meanwhile, the Ohira–Bestmann procedure, in which DAMP is produced in situ from dimethyl-1-diazo-2-oxopropylphosphonate, has become the most popular method of transforming an aldehyde into the corresponding alkyne under mildly basic reaction condition (K2CO3/MeOH) (Eq. 2).34,35) However, using this method, ketones cannot be transformed into internal alkynes, and α,β-unsaturated aldehydes do not yield enynes.36)

Chart 12. [1,2]-Migration and [1,5]-C–H Insertion Reactions of Alkylidene Carbenes

Chart 13. Current Procedures for Homologous Alkyne Synthesis from Carbonyl Compounds
In continuation of our program of using CPs, we have recently reported novel synthetic methods for homologous alkynes (45)13) or five-membered unsaturated cyclic compounds (47)37) from carbonyl compounds (4 and 5) based on reactions of CPs (3) with trimethylsilyl azide (TMSN3) in the presence of Bu2SnO as catalyst (the Wittenberger method),38) as shown in Chart 14. In this two-step transformation, CPs (3) may form tetrazolylphosphates (48), which subsequently undergo successive fragmentation to generate alkylidene carbenes (44 and 46) that then undergo [1,2]-migration or [1,5]-C–H insertions to produce homologous alkynes (45) or five-membered cyclic compounds (47), respectively. The scope of these reactions, which occur under neutral conditions, could be extended towards a variety of alkynes and cyclopentene products that are not usually accessible from the corresponding carbonyl compounds via the lithium trimethylsilyldiazomethane [TMSC(Li)N2] procedure39–41) used currently in organic synthesis. In addition, the TMSC(Li)N2 procedure requires basic conditions.

Chart 14. Proposed Mechanism for Formation of Alkynes and Cyclopentenes Mediated by Tetrazole-Fragmentation from CPs
We first found that reaction of benzylic CPs 50 with sodium azide in the presence of Et3N·HCl gave α-azidotetrazoles (ATs) 51. Treatment of ATs 51 thus obtained under microwave (MW) irradiation gave the expected alkynes 52, as shown in Chart 15.13)

Chart 15. Transformation of CPs 50 into Alkynes 52 under MW Conditions
The procedure outlined above still requires isolation of unstable ATs and relatively harsh conditions in the MW reactor. In the search for a milder variant, it was found that CPs 50 could be transformed directly into the corresponding alkyne 52 under Wittenberger conditions,38) i.e., by using 1 equiv. of TMSN3 in the presence of a catalytic amount of Bu2SnO (0.1 eq.) at reflux in toluene, as shown in Chart 16.

Chart 16. Transformation of Ketones 53 into Alkynes 52 via CPs 50
Thus, many ketones 53 can be transformed into the corresponding alkynes 52 in moderate to high yields using this method.13) The potential of the reaction methodology has been further explored by applying it to the synthesis of a selective mGlu5 receptor antagonist, 2-methyl-6-(phenylethynyl)pyridine (54: MPEP).42) Starting from 6-methylpicolinaldehyde, MPEP was obtained in 68% overall yield over four steps, as shown in Chart 17.13)

Chart 17. Synthesis of the mGlu5 Receptor Antagonist, MPEP
Interestingly, β-ketoester 55, which possesses readily enolizable α-protons, could also be converted into the corresponding alkyne 56 (68%) by the present method (Chart 18). In contrast, the reaction of 55 under Shioiri conditions [TMSCHN2/lithium diisopropylamide (LDA)]39) only resulted in the unreacted starting material.

Chart 18. Transformation of β-Ketoester 55 to Its Corresponding Acetylenic Compound 56
Colvin and Hamill’s original procedure used either TMSCHN2 or DAMP, and its modifications cannot be applied to dialkyl ketones.30–32) Furthermore, the Ohira–Bestmann reaction of ketones does not afford alkynes; instead, enol ethers are formed.34,35) As shown in Chart 19, the method developed in this work furnished the corresponding 1,6-diphenylhexen-3-yne (58) from 1,5-diphenylpentan-3-one (57) in 77% overall yield (Eq. 1). In contrast, the Ohira–Bestmann reaction of 57 gave methyl enol ether 59 in 40% yield (Eq. 2),34,35) whereas the Shioiri procedure yielded allenylsilane 60 (31%),43) and a homologous aldehyde 61 (23%),40) as well as unreacted 57 (46%) (Eq. 3). The formation of allenylsilanes or homologous aldehydes from ketones using TMSCHN2 has been reported independently by the Lee43) and Shioiri groups.40) These results indicate the versatility of our method.

Chart 19. Reactivity of Dialkyl Ketone 57 under Three Different Conditions
The present method was successfully extended to the transformation of aldehydes 62 into homologous terminal alkynes 64, as shown in Table 5. In our studies on the synthesis of novel triazole-containing RNA or DNA,44) we sought access to alkynes 64a from β-D-ribofuranosyl aldehydes 62a. The reaction of CP 63a with NaN3 (3 eq.) and Et3N·HCl (3 eq.) gave β-ribofuranosyl alkyne 64a in 68% yield (method A) (Table 5, entry 1). Alternatively, treatment of 62a with TMSN3 (1 eq.) in the presence of Bu2SnO (0.1 eq.) gave 64a in 47% yield (method B) (entry 1). Thus, method A is favorable for the preparation of homologous alkyne from the corresponding aldehyde. Both methods A and B may be employed equally for primary aldehydes (62b, entry 2), but method A is slightly more useful for aldehydes 62b (entry 2). Conversely, in the case of the aromatic and α,β-unsaturated aldehyde CPs 62c and 62d, respectively, method B is favorable, providing alkynes 64c and 64d in 71% and 80% yields, respectively (entries 3 and 4). The reason for the preference towards either method A or B depending on the aldehyde is unknown at present.
Table 5. Transformation of Aldehydes into Terminal Aldehydes
via CP
a) Isolated yield. b) Reflux, 0.5 h. c) TMSN3 (3 eq.), Bu2SnO (0.1 eq.), reflux, 0.5 h.
As shown in Chart 20, the transformation of aldehyde 62e into alkyne β-64e in overall 99% yield is of particular interest, because the Ohira–Bestmann reaction and Shioiri modification procedures gave only inseparable 5 : 2 and 9 : 1 epimeric mixtures of β-64e and α-64e, respectively.44) The formation of the epimeric mixtures probably occurs because of the extraction of α-acidic proton of 62e followed by β-elimination under basic conditions.

Chart 20. Reactivity of β-D-Ribofuranosyl C1-Carbaldehyde (62e) under Different Conditions
As mentioned before, the Ohira–Bestmann reaction does not give enynes from α,β-unsaturated aldehydes.34,35) In contrast, it is possible to synthesize the corresponding enyne 64d from cinnamaldehyde (62d) in 78% overall yield following the method described in this work (Chart 21). Furthermore, the Ohira-Bestmann reaction using 62d as substrate gave (1-methoxybut-3-nyl)benzene (65) in 10% yield, together with dimethyl acetal (66, 67%).

Chart 21. Reactivity of Cinnamaldehyde 62d Using the Present and Ohira–Bestmann Methods
The mechanism of Bu2SnO-catalyzed cycloaddition of TMSN3 on nitriles has been studied in detail by Kappe and colleagues.45) It was demonstrated that the active catalytic species is Bu2Sn(OTMS)N3 (67), and that regeneration of this catalyst occurs through SN2 displacement at the silicon atom followed by fast ligand exchange at the tin atom. A plausible mechanism that would account for the formation of alkynes 45 from CPs 3 is shown in Chart 22. The proposed mechanism also indicates that the C1 unit of homologous alkynes 45 arises from the CN-carbon of DEPC.

Chart 22. Proposed Mechanism for the Formation of Alkynes 45 Mediated on Tetrazole-Fragmentation from CPs 3
Synthesis of five-membered unsaturated cyclic compounds from ketones by [1,5]-C–H insertion of alkylidene carbenes was carried out using (MeO)2P(O)CHN2 or TMSCHN2 in the presence of a strong base.28,29) Methyl alkyl ketones 70 with lower [1,2]-migratory aptitudes were thus chosen as substrates for the formation of various five-membered compounds, as summarized in Table 6. In the case of methyl ketones 70a–d (entries 1–4), cyclopentene 72a, 2,5-dihydrofuran 72b, and 2,5-dihydropyrroles 72c, and 72d were readily obtained in 66%, 59%, 75%, and 89% overall yields in two steps. Furthermore, the present method was successfully applied to the conversion of (4S)-2,2-dimethyl-4-(3-oxobutyl)-1,3-dioxolane (70d) into spiro[4.4]nonene (72d, 89% overall yield, entry 4), which is a versatile intermediate in the total synthesis of (−)-malyngolide46) or (−)-frontalin47) as reported by Ohira and co-workers. They employed Shioiri conditions (TMSCHN2/n-BuLi)39–41) or the Seyferth–Gilbert procedure (DAMP/t-BuOK)30–32) for the preparation of 72d from ketone 70d, obtaining the products in 73 and 68% yields, respectively. The results summarized in Table 6 indicate that our method is preferable to existing methods.
Table 6. Transformation of Methyl Alkyl Ketones
70 into Five-Membered Products
72 via CPs
71a) DEPC (3.0 eq), LiCN (3.0 eq.), 1 h, rt; b) TMSN3 (1.0 eq.), Bu2SnO (0.1 eq.), 2 h, reflux; c) TMSN3 (2.0 eq), Bu2SnO (0.2 eq), 4 h, reflux; d) 2 steps, overall yield from 70 by our method; e) Shioiri procedure using TMSCHN2/BuLi; f) Gilbert procedure using DAMP/tert-BuOK; g) Figures in parentheses refer to the results of other methods.
The present method could further undergo [1,5]-O-SiMe3 bond insertion41) to generate 5-trialkylsilyl-2,3-dihydrofurans 75a (77%) and 75b (81%) from the corresponding CPs 74a (R = TMS) and 74b (R = TBDMS), respectively (Chart 23).

Chart 23. Formation of 5-Alkylsilyl-2,3-dihydrofurans Using the Present Method
Furthermore, the 16-membered cyclic ketone 76 yielded bicyclo[12.2.1]heptadecene (77, 59%) via a CP together with cyclic alkyne homolog 78 (16%), in which [1,5]-C–H insertion mediated by alkylidene carbene from CP was preferred to the formation of cyclic alkyne homolog 78, as shown in Eq. 1 of Chart 24. In contrast, Lee and co-workers reported that the reaction of small- to medium-sized cyclic ketones (76, n = 4–14) with TMSC(Li)N2 led to allenylsilanes (79, 10–81%) as per Eq. 2.43)

Chart 24. Transformation of Cyclic Ketone 76 into Bicyclic Compound 77
Notably, β-keto ester 80, which possesses readily enolizable α-protons, could be converted into cyclopentenyl acetate 81 in 95% overall yield using the present method (Chart 25, Eq. 1). In contrast, the reaction of 80 under Shioiri conditions (TMSCH2/LDA) only resulted in the unreacted starting material (80), as per Eq. 2.

Chart 25. Transformation of β-Keto Ester 80 into Cyclopentenyl Acetate 81
(−)-Neplanocin A (NPA, 91) is a naturally occurring carbocyclic nucleoside49) (Chart 26). NPA and other natural analogs have received considerable attention because of their interesting biological properties, such as their potent antiviral and antitumor properties.50) NPA itself is cytotoxic to host cells,51) and this may account for its reduced therapeutic potency. Therefore, the chemical synthesis and structural modification of NPA have been investigated extensively.50) Among them, protected cyclopentenone 89 and cyclopentenyl tetrol 90a have been widely used as synthetic precursors not only for NPA, but also its analogs.52–54) Based on these reports and to demonstrate the synthetic utility of the alkylidene carbenes generated from CPs, we attempted to apply them to the synthesis of NPA (−)-91 and its synthetic precursors 89 and 90a from ketone 83,53) as shown in Chart 26.

Chart 26. Synthesis of (−)-Neplanocin A from CP 84
Cyanophosphorylation of ketone 83 easily afforded CP 84 in 95% yield, The reaction of CP 84 with TMSN3/Bu2SnO afforded an inseparable mixture of epimeric cyclopentenes 86αβ, which was the result of the C–H insertion reaction of alkylidene-carbene 85, along with unexpected dihydropyran 87.53) After the mixture was treated with tetrabutylammonium fluoride (TBAF) to remove the TBDMS group, deprotected cyclopentenes 88αβ (88α/88β = 1/3) and dihydropyran 87 were obtained in 65 and 20% yields, respectively, from CP 84. As illustrated in Fig. 2, the formation of 87 may have been caused by [1,6]-O-Si bond insertion of the carbene 85a or 1,2-TBDMS shift of ylide 85b,54) as opposed to the usual [1,5]-O-Si insertion that generates dihydrofuran derivatives.28,41)
4. Schematic Diagram on Applicability of CPs
A number of applications of CPs in organic synthesis are being developed continuously by researchers at home and abroad since the first author’s report on CPs. These studies are described in detail in the recent review of DEPC,1) and the applicability of CPs in organic syntheses may be summarized in Figs. 3 and 4.1)
5. [3,3]Sigmatropic Rearrangement of Cyclic Thionocarbonates of Medium Ring-Size55)
5.1. Sigmatropic Rearrangement of Medium-Sized ThionocarbonatesThe exclusive E-selectivity of the double bond created by [3,3]sigmatropic rearrangement has been extensively used for natural product synthesis. However, few synthetic studies on the opposite (Z)-selective [3,3]sigmatropic rearrangement have so far been reported, probably due to lack of suitable methodology. Meanwhile, medium-ring compounds (those having a ring size in the range 8 to 11) are becoming increasingly important in organic chemistry, because they are discovered in an ever-growing number of natural products.56) In this context, we found that the [3,3]sigmatropic ring expansion of allylic 8-membered thionocarbonates induced the highly stereoselective synthesis of either Z or E olefins in 10-membered thiolcarbonates. In this chapter, we describe our results on the [3,3]sigmatropic rearrangement of cyclic thionocarbonates of medium ring-size and their application to the synthesis of natural products.
In our study of the formation of the 8-membered thionocarbonates,57) we found spontaneous ring expansion of the cyclic thionocarbonates via [3,3]sigmatropic rearrangements.58,59) When a dry THF solution of (TMS)2NNa was rapidly added to a THF solution of diol monothionocarbonate 92 at r.t., the reaction went to completion immediately to give a 10-membered thiolcarbonate 94 containing a (Z)-double bond in 78% yield58) (Chart 27).

Chart 27. Ring Expansion via [3,3]Sigmatropic Rearrangement of Cyclic Thionocarbonate 93
The formation of a (Z)-double bond in 94 can be rationalized as follows. In the [3,3]sigmatropic rearrangement, 93 can adopt two possible conformations (Tc and Tt) as transition states, as shown in Chart 28. The Tc (n ≤ 4) with 1,3-diaxial interaction would lead to the observed (Z)-olefin. In the case of 93 (n = 4), the transition state Tt bearing the tethered chain in a trans relationship should be excluded because of the strain incurred in the four-carbon tether during alignment of the thiocarbonyl group and the double bond for the [3,3]sigmatropic ring expansion. In the case of larger thionocarbonates (n ≥ 5), the ring size would be sufficiently large to allow the latter conformation (Tt).59) Relationship between the ring size of cyclic thionocarbonates and the geometries of the products was also clarified by our experiments.59)

Chart 28. Relationship between Ring Size of Cyclic Thionocarbonates and Z or E Geometries of Products
Whether the introduction of an alkyl group into the allylic system of the substrates (monothionocarbonates) influences the geometry of the double bond created in 10-membered thiocarbonates was investigated in detail.60) Among them, reaction of diol monothionocarbonate E-95 having a butyl group with (TMS)2NNa went to completion immediately to give a (Z)-10-membered thiolcarbonate (Z)-97 in 88% yield61) (Chart 29, Eq. 1). Meanwhile, the same treatment of (Z)-95 having a butyl group provided an (E)-10-membered thiolcarbonate (E)-97 in 78% yield (Eq. 2).61) Spectroscopic analysis showed (Z)- and (E)-97 to have 100% isomeric purity.61) The Z-selectivity of (Z)-97 can be explained by chair-like transition state TC, whereas the boat-like transition state TB leads to the opposite geometry, as illustrated in Fig. 5. Conformational energy calculations supported the validity of the chair-like and boat-like transition states TC and TB in the [3,3]sigmatropic ring expansion of cyclic thionocarbonates 96.62)

Chart 29. Highly Stereoselective Synthesis of Z- or E-Double Bonds with Conformational Control in [3,3]Sigmatropic Ring Expansion of 8-Membered Thionocarbonates 96
The yellow scale, Aonidiella citrina, is a severe pest of ornamental plants and citrus fruit in California and Japan. The sexual pheromone of the yellow scale (S)-(E)-6-isopropyl-3,9-dimethyl-5,8-decadienyl acetate (104) has been synthesized by several groups. The major difficulty in the preparation of this compound is stereoselective synthesis of the double bond bearing the isopropyl group. In continuation of the study on the [3,3]sigmatropic ring expansion of medium-sized cyclic thionocarbonates, we achieved unique and stereoselective synthesis of this yellow scale pheromone from chiral aldehyde (+)-99, as shown in Chart 30.65)

Chart 30. Synthesis of (−)-Yellow Scale Pheromone via [3,3]Sigmatropic Ring Expansion of 8-Membered Thionocarbonate Containing A Diene Moiety
In this synthetic study,63–65) we developed a method of direct one-pot conversion to the key 10-membered thiolcarbonate 103 from chiral aldehyde 99, and (−)-yellow scale pheromone 104 was achieved via 103.65) This conversion consists of four reactions, as illustrated in Chart 30: i) Generation of dienyllithium 100 in pentane. ii) Addition of 100 to the chiral aldehyde 99. iii) Cyclization of the lithium alkoxide 101 to 8-membered thionocarbonate 102. iv) The [3,3]sigmatropic ring expansion to the 10-membered product 103. It should be noted that THF is an indispensable solvent for promoting the steps iii and iv. Reductive removal of the SCO moiety in 103 thus prepared by lithium p,p′-di-tert-butylbiphenylide (LDBB)-hexamethylphosphoric triamide (HMPA) followed by acetylation finally afforded (−)-yellow scale pheromone 104.
5.4. Synthesis of Medium-Sized Heterocyclic Allenes and Its Synthetic Application66,67)Alternatively, treatment of acetylene diol monothionocarbonates 105 with (TMS)2NLi gave medium-sized heterocyclic allenes 107 via spontaneous [3,3]sigmatropic ring expansion of alkynyl cyclic thionocarbonates 106, as shown in Chart 31.67)

Chart 31. Synthesis of Medium-Sized Heterocyclic Allenes
The formation of the medium-sized heterocyclic allenes was applied to the synthesis of a racemic form (±)-115 of (R)-(−)-methyl 8-hydroxy-5,6-octadienoate, which is a strong antifungal constituent in the leaves of a deciduous tree native to Japan, Sapium japonicum, as shown in Chart 32.66) This synthesis also features a novel application of the SmI2-HMPA reduction to a key intermediate, 10-membered thiol carbonate 111. Reaction of diol monothionocarbonate 109, prepared from aldehyde 108, with (TMS)2NLi afforded a 10-membered allene 111 in 85% yield via the [3,3]sigmatropic ring expansion of a cyclic thionocarbonate 110. Subsequent treatment of 111 with SmI2-HMPA reduction afforded an allenic alcohol 112 in 78% yield with the liberation of SCO. The bifunctional compound 112 was converted to (±)-115 via four steps.

Chart 32. Synthesis of Antifungal Constituent of Sapium Japonicum
Decreasing the ring size of cyclic allenes results in deviation of both the normal C=C=C linearity and the orthogonality of dihedral angle. Treatment of diol monothionocarbonate 116 having a tert-butyl alkyl group with (TMS)2NLi in THF at r.t. afforded a 6-membered cyclic thionocarbonate 11767) (Chart 33, Eq. 1). Rearrangement of 117 as expected proceeds upon refluxing in benzene solution for 1.5 h to give a pure 8-membered heterocyclic allene 118. The structure of 118 was estimated from modified neglect of diatomic overlap (MNDO) calculation results (Eq. 2).67) The bond angle on the sp carbon, C1–C2–C3, is decreased from linearity to 170.1°. The dihedral angle CH3–C1–C3-t-Bu, is twisted (64.3o) from vertical geometry

Chart 33. Synthesis of Strained 8-Membered Heterocyclic Allene 118 and Its Derivative
We then examined the behaviour of the 8-membered cyclic allenes 118 for a base or acid.67) Intriguing was the formation of 2H-thiopyran derivative 121 by treatment of 118 with 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU), inducing isomerization of the double bond of 118 and elimination of CO2 from the resulting cyclic diene 120 (Chart 33, Eq. 3). However, no change was observed by treatment of 118 with p-toluenesulfonic acid in THF.
6. Synthetic Study of Imidazole C-Nucleosides and Their Application to Probing Catalytic Mechanisms of Ribozymes68,69)
6.1. Stereoselective Synthesis of C-4 Linked β-Imidazole C-RibonucleosidesC-Ribonucleosides have attracted much attention in view of their remarkable antitumor and antiviral activities.70–72) Azoles are biologically important heterocyclic compounds, and a variety of useful therapeutic agents containing the imidazole moiety have been developed.73,74) Meanwhile, the synthetic method of imidazole C-nucleosides, in general, is limited and requires multiple steps.70–72) We thus directed our attention to synthesizing 4(5)-(β-D-ribofuranosyl)imidazole (127) having the imidazole in place of nucleo-base, as shown in Chart 34.75,76)

Chart 34. Stereoselective Synthesis of C-4 Linked β-Imidazole C-Nucleosides
The reaction of tribenzyl-D-ribose 122 with lithium salt 123 of bis-protected imidazole gave inseparable epimeric mixture RS-124, as illustrated in Chart 34. Hydrolysis of RS-124 in refluxing 1.5 N HCl afforded a 1 : 1 mixture RS-125 of diols having an unsubstituted imidazole. Treatment of the mixture RS-125 with N,N,N′,N′-tetramethylazodicarboxamide (TMAD)-Bu3P at rt in benzene exclusively produced the desired β-anomer (β-126) in 92%, together with a small amount of α-anomer (α-126, 3.5%). The ratio of β- and α-anomer was 26.3 : 1. Debenzylation of β-126 was carried out to achieve the synthesis of C-4 linked β-imidazole C-ribonucleoside β-127 in four steps and 87% overall yield from the starting 122.
Importantly, it is possible to synthesize the β-anomer without separation of the isomers (R-125 and S-125) or stereoselective synthesis of the R-isomer which can provide β-anomer through intramolecular SN2 reaction under standard Mitsunobu reaction. Accordingly, the unsubstituted-imidazole moiety is indispensable for exclusive formation of β-anomer. The β-stereoselectivity in this reaction can be rationalized as in Chart 35. Reaction of TMAD-Bu3P adduct with R-125 forms a zwitterion R-128. Preferential elimination of Bu3P=O from 128 leads to diazafulvene 129a. Spontaneous cyclization of 129a gives β-126. Although S-125 similarly leads to active species 129b, it exclusively gives the β-anomer via rotamer 129a being thermodynamically more stable. The remarkable stereoselectivity (β/α = 26/1) of the β-ribofuranosylimidazole is facilitated by electron repulsion in 129b.

Chart 35. Mechanistic Consideration on β-Stereoselective Glycosidation
The stereoselective synthetic method of C-nucleosides via diazafulvene intermediates was further applied to synthesis of various C-nucleosides: β-ribofuranosylimidazoles exhibiting antiulcer activity77); α-L-arabinofuranosylimidazoles78); tetrahydrofuranylimidazoles79,80); α-D-xylofuranosylimidazoles81); trans- or cis-4(5)-(5-aminomethyltetrahydrofuran-2-yl)imidazoles for histamine H4-ligands82); and pyrazole C-nucleosides.83)
6.2. Synthesis of Novel C4-Linked Imidazole Ribonucleoside Phosphoramidites for Probing General Acid and Base Catalysis in Ribozymes69)The VS ribozyme is the largest of a group of nucleolytic ribozymes that include hammerhead, hairpin and HDV ribozymes, and catalyzes the site-specific cleavage of a phosphodiester linkage to generate products containing 2′,3′-cyclic phosphate and 5′-OH termini.84) The VS ribozyme is the only member of the class of nucleolytic ribozymes for which there is no known crystal structure.84) Lilley and colleagues recently indicated that the A730 loop of VS ribozyme is an important component of the active site in general acid and base catalysis85) (Fig. 6). In particular, the adenine (A756) in the A730 loop is a critical base in the cleavage,86) because sequence variants at the position 756 are especially impaired in the cleavage and ligation activity. In our collaborative studies with Lilley and colleagues, a trans-acting VS ribozyme, where a three-piece ribozyme-substrate system was used, have been designed, as illustrated in Fig. 6.
Given that imidazole with a pKa of 7.1 is both a good donor and acceptor of proton, synthesis of an imidazole C-nucleoside would allow incorporation at the desired position to probe general acid and base catalysis. Therefore, synthetic study of the tribenzyl-β-ribofuranosylimidazole β-126 led us to the development of a new chemical strategy for determining the role of acid-base catalysis in a ribozyme, in which C4-linked imidazole was placed into A756 of VS ribozyme covalently as a pseudonucleobase, as illustrated in Fig. 7.87,88)
In this study, a novel C4-linked imidazole 5′-O-dimethoxytrityl (DMT)-2′-O-tert-butyldimethylsilyl (TBDMS)-3′-O-cyanoethyldiisopropylphosphoramidite 130 was designed as a synthetic unit of the A756Imz VS ribozyme, and synthesized starting from β-126, as shown in Chart 36. Investigation of protecting groups for the imidazole-N first indicated that pivaloyloxymethyl (POM) was adequate as an N-protecting group for the imidazole nucleoside β-126 which could be readily removed under mild basic condition [aqueous ammonia–MeOH (1 : 3, v/v), r.t., 3 h]. Therefore, treatment of β-126 with POMCl followed by Pd(OH)2-C/cyclohexene produced N-POM-imidazole C-nucleoside 131 (Chart 36). DM-tritylation of 131 afforded 5′-O-DMT-derivative 132. Treatment of 132 with TBDMSOTf /Py led to an inseparable 1 : 1 mixture 133ab (52%) of 2′-O-TBDMS and 3′-O-TBDMS isomers, together with a 2′-,3′-bis-O-substituted derivative 134 (17%). The mixture of 133ab was subjected to phosphitylation with 2-cyanoethyl tetraisopropylphosphodiamidite to yield the desired 3′-phosphoramidite (PA) 130 (31%) and 2′-PA 135 (25%). However, the synthetic route of PA 130 led to problems of instability against the purification on silica gel column chromatography and low overall yield (<5%).
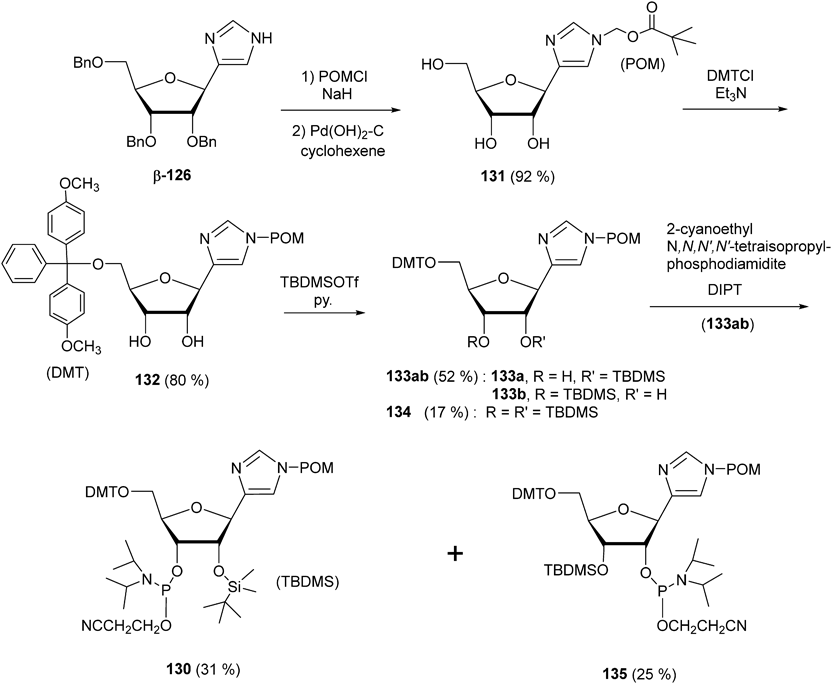
Chart 36. Synthesis of C4-Linked Imidazole Ribonucleoside PA 130
The imidazole PA 130 thus prepared was subjected to a TBDMS approach of RNA automated synthesis to provide an imidazole-substituted VS ribozyme (A756Imz) in an average 99% coupling yield89) (Fig. 7). The A756Imz catalyzed the almost-complete cleavage of a substrate stem-loop at the correct position, as shown in Fig. 8.89) Virtually complete cleavage has been obtained, with rates of 0.01 min−1. The result powerfully supported a direct role of the nucleobase at position A756 in the chemistry of natural VS ribozyme.89)
In another study, PA 130 was also used to replace G8 in the hairpin ribozyme with the imidazole base. The modified hairpin ribozyme (G8Imz) catalyzed both cleavage and ligation reactions, with a pH dependence consistent with a role of G8 in general acid-base catalysis.90)
6.3. Accurate Molecular Weight Measurements of Nucleoside PAs: A Suitable Matrix of Mass Spectrometry91,92)Nucleoside PAs are the most widely used building blocks in contemporary solid-phase synthesis of oligonucleotides. The novel imidazole PA 130 with POM group, which is easily removed under basic conditions and compatible with TBDMS synthesis of RNA, was first synthesized as a key ribonucleoside variant, as mentioned in chapter 6.2. However, in course of analytical investigation of 130, we encountered difficulty in mass spectrometric characterization of 130, which decomposed even on a standard TLC plate (Merck 60 F254). After many investigations, a collaborative study with Fujitake clarified that the accurate molecular weight (MW) measurements of such molecules, which contain acid-labile moieties, may be easily determined by mass spectrometry using a new matrix system, triethanolamine (TEOA)-NaCl, on liquid secondary ion mass spectrometry (LSIMS) equipped with a double-focusing mass spectrometer, as illustrated in Fig. 9.93) In addition, this matrix has an advantage of permitting use on the alternative ionization method, FAB.94) The present method measures rapidly and easily the accurate MWs of various PAs as adduct ions [M + Na]+ with average mass error smaller than 0.4 ppm, allowing the formulas of various PAs in place of elemental analysis. The sodium-cationized molecule of PA130 appeared at m/z 953.4625 in the externally calibrated spectrum (polyethylene glycol) by high-resolution (HR)-MS and the error was only 0.4 ppm from the expected mass number, as shown in Fig. 10.93) Furthermore, all of more than 50 PAs investigated, which bear a variety of functional groups, gave sodium-cationized molecules using the present method.93) These observations successfully demonstrate the generality and potential of the method. Hence, the proposed method is a powerful tool for MS identification of PAs or fragile molecules.
6.4. Efficient Synthesis of 2′-O-Cyanoethylated Imidazole C-Ribonucleoside PAsWe have described previously that a 2-O-TBDMS C4-linked imidazole ribonucleoside PA 130 can be applied as a key building block to investigate the role of general acid-base catalysis in VS and hairpin ribozyme RNA using solid-phase TBDMS chemistry implemented on an automated synthesizer. However, there is a serious hindrance to the general application of this approach. As shown in Chart 36, the synthetic route of PA 130 lacks the selective introduction of TBDMS group at 2′-hydroxy group of diol intermediate 132 owing to easy 2′-3′ TBDMS-migration, leading to the inseparable 1 : 1 mixture 133ab. Thus, when the mixture 133ab was subjected to phosphitylation, PA 130 was only obtained in poor yield (31%) together with 2′-PA 135 (25%). Moreover, great care has to be taken for purification and isolation of 130 because of its acid-labile character and contamination by phosphorus residues. As a result, the overall yield of 130 is <5% through 8 steps from the starting tribenzyl D-ribose 122. In our systematic studies on the catalytic mechanism on ribozymes, we developed a variant PA 140, in which the cyanoethyl (CE) group was used as the smallest available protecting group for the 2′-hydroxy function, as illustrated in Chart 37.95) It is particularly significant that a combination of CE and POM protecting groups inhibited 2′-3′ migration between 2′- and 3′-hydroxy groups and provided additional stability for purification of the PA 140. The total yield of 140 was 41% in ten steps from the commercially-available tribenzyl D-ribose 122, demonstrating the effectiveness of this approach (Chart 37). This has resulted in a marked improvement of yield compared with unstable 2′-O-TBDMS PA 130.95) The new synthon PA 140 enables the imidazole moiety to be incorporated into any RNA sequence in principle, and has been used as a practical building block for probing the catalytic mechanism of a ribozyme.

Chart 37. Synthesis of 2′-O-Cyanoethyl Imidazole C-Nucleoside PA 140
In our studies of the catalytic mechanism of VS ribozyme, Lilley identified a guanine (G638) in the substrate loop of VS ribozyme as a second important nucleobase for general acid-base catalysis96) (Fig. 6). However, when we substituted C0-linked imidazole nucleoside at position 638 using PA 130, the modified VS ribozyme (G638C0Imz) exhibited virtually no restoration of cleavage activity.96) This result suggests that the environment at the position 638 was more constrained, preventing the structural rearrangement that would be required to bring the imidazole base to the position normally occupied by the guanine N1. It might be anticipated that the manner of linking the imidazole group to the ribose would affect its ability to function in general acid base catalysis. If the linker is too short then its ring N cannot be superimposed with that of N1 of adenine or guanine. On the other hand, if it is too long then it may spend too little time in position required for its potential catalytic function. Furthermore, molecular graphics shows that the ring N atom of imidazole C2-nucleoside and N1 of guanine can readily be superimposed, whereas these atoms are separated by ≥2 Å for the C0-nucleoside in the absence of significant distortion.96) Consequently, we designed and synthesized a two-carbon-elongated homologue (Imz-C2-PA, 149) that may be a better structural mimic of purine nucleobase.96) As shown in Chart 38, the synthesis of PA 149, in which POM and 2-CE groups were used to protect imidazole-N and 2′-hydroxy function, was successfully achieved in 13 steps and 23% overall yield from starting tribenzyl D-ribose 122.

Chart 38. Synthesis of ICN-C2-PA 149
The C2-imidazole nucleoside, a flexible structural mimic of a purine nucleobase, was successfully incorporated using the Imz-C2-PA 149 into position 638 of VS ribozyme through 2′-TBDMS chemistry to study the role of G638 in general acid-base catalysis.96)
It is interesting to note that the cleavage rate of G638C2Imz was 15-fold greater than that achieved with the ribozyme (G638C0Imz) containing a C4-linked imidazole at the same position, as shown in Table 7.96)
Table 7. Comparison of Cleavage Rates between G638C
2Imz and G638C
0Imz
| Rates/min−1 natural | G638C2Imz | G638C0 Imz | C2Imz/C0 Imz |
|---|
| 10 mM Mg2+ pH 8 |
| −Rz | | 6 × 10−5 | 5 × 10−5 | |
| +1 µM Rz | 0.61 | 0.001 | 7 × 10−5 | 14 |
| 200 mM Mg2+ pH 6.5 |
| −Rz | | 1 × 10−5 | 2 × 10−5 | |
| +1 µM Rz | 6.2 | 0.008 | 5 × 10−4 | 15 |
From these results, G638 is one of the nucleobases proposed to act in general acid-base catalysis in concert with A756 in the catalytic mechanism of the VS ribozyme.96) The chemical mechanism appears to involve general acid-base catalysis via the combination of G638 and A756 nucleobases which are juxtaposed with the scissile phosphate, as illustrated in Fig. 11.84)
The VS ribozyme is the only member of the class of nucleolytic ribozymes for which there is no crystal structure, and mechanistic investigation cannot be guided by structural data.84) Because the whole series of ribose-(CH2)n-Imz PAs (n = 0–3) could make an interesting series of compounds for systematically tracing out the geometry of the active site of ribozyme, Imz-C1- and C3-PAs 150 and 151 were prepared (Fig. 12A).97) Furthermore, starting from tetrazole(Tez)-C0- and C2-PAs 152 and 153 (Fig. 12B), C5-linked C0- and C2-tetrazoles were incorporated successfully into the VS ribozyme substrate to determine more specifically which nucleobases of the ribozymes function as the acid or base.98) Ribose-(CH2)n-Imz or Tez species can, in principle, be incorporated at any site of RNA sequences through chemical synthesis. In addition, Imz-C0- and C2-deoxyribonucleoside phosphoramidites (dPAs, Fig. 12C) are successfully incorporated into the sequence of a 15-nt DNA, and the abilities of one or two imidazoles to pair with different bases are investigated through thermal melting (Tm) experiments on the resulting DNA duplexes.99)
7. Novel Synthetic Methods Using Se or Sn Atoms towards Histamine H3 Receptor Ligands69,100)
7.1. Synthesis of Tetrahydrofuranylimidazoles Using a PhSe Group and Its Application to New Histamine H3 Receptor Agonist, ImifuramineHistamine receptors are a class of G protein-coupled receptors with histamine as their endogenous ligand. The histamine H1 receptor (H1R) is responsible for inflammation and allergic reactions, whereas the histamine H2 receptor (H2R) is responsible for the mechanism of gastric acid secretion. Both H1R and H2R antagonists are nowadays in widespread clinical use. The histamine H3 receptors (H3R), on the other hand, exist at the varicosities and endings of histaminergic fibers in the brain and modulate the synthesis and release of histamine as an autoreceptor, as illustrated in Fig. 13.101) Moreover, H3Rs are heteroreceptors that modulate the release of a number of different neurotransmitters. Therefore, histamine neurons play an important role in arousal, learning, and memory mechanisms, working together with other neuromodulatory systems. H3R-antagonists are expected to be potential drugs for memory degenerative disorders such as Alzheimer’s disease. This type of receptor can be also found in many peripheral tissues. Since H3R-Agonist (R)-α-methylhistamine, imetit, and imepip were shown to possess inhibitory actions against airway smooth muscle contraction, H3R-agonists are regarded as a target for new therapeutics of bronchial asthma.102)
The most recently identified histamine H4 receptor (H4R) is mainly expressed in hematopoietic cells such as mast cells, basophils, eosinophils, T-cells, and dendritic cells, as shown in Fig. 14.103) It is therefore believed to play an important role in inflammation and immune responses, with possible applications in diseases such as inflammatory bowel disease, allergic asthma, and pruritus.103)
A reoccurring issue in the development of H3R antagonists is selectivity between H3R and H4R. These two receptors share the highest homology within the histamine receptor family (31% overall, 54% in the transmembrane region) (Fig. 14). Indeed, several classical H3R antagonists, such as thioperamide and clobenpropit,104) also have significant affinity for H4R. To investigate the physiological and pathophysiological roles of H3R and H4R, the development of a range of selective H3R ligands is crucial.
Theoretical calculations of histamine or some H3R-agonists have predicted the importance of an intramolecular hydrogen-bonding between the cationic primary amine and the N atom of the imidazole, leading to a folded model A, and an extended model B as bioactive conformations of H3R-agonists was also reported105,106) (Fig. 15). However, they have not been experimentally demonstrated.
From the β-stereoselective synthesis of C-4 linked imidazole nucleosides, we envisioned that, while the cis-isomer 156 of 4(5)-[5-(aminomethyl)tetrahydrofuran-2-yl]imidazole could adopt a folded conformation through intramolecular hydrogen-bonding, the trans-isomer 157 would form the extended one (Fig. 16).
Therefore, we synthesized four isomers of novel THF derivatives 156 and 157 using a synthetic method characterized by use of a phenylselenyl (PhSe) group for the formation of the THF ring.107,108) Introduction of a PhSe group into lactone (+)-158 prepared from L-glutamic acid gave C2α-and C2β-selenolactone 160α (52%) and 160β (30%) (Chart 39). Both of which were used as substrates to prepare key intermediates 164α and 164β. Reduction of the major isomer 160α with DIBAL followed by an addition of 5-lithioimidazole to the resulting lactol 162S (73%) with a C1′S configuration, together with C1′ epimer 162R (10%). The anti-selectivity for 162S may be account for by a chelation-cyclic model as indicated in Fig. 17.

Chart 39. Synthesis of 1′,2′-Anti-diol 162S
Deprotection of 162S under reflux in HCl-THF afforded easily separable β- and α-nucleoside derivatives 164β (42%) and 164α (56%) (Fig. 18). The formation of 164β and 164α can be reasonably rationalized as shown in Fig. 18. The reaction of 162S with HCl generates olefin 163 by a trans-stereospecific elimination of PhSeCl. Recombination of 163 and PhSeCl gives β- and α-anomers 164β and 164α through selenium-induced cyclization at both faces of double bond of 163. Similar treatment of 162R provided 164β and 164α. This synthetic method for the preparation of C-nucleoside derivatives using a combination of the elimination of PhSeCl and selenocyclization has not been reported.108)
Ethoxycarbonylation of the α-anomer 164α gave 165α which was then converted into 166α by free radical deselenylation (Chart 40). Debenzylation of 166α and subsequent phthaloylimination afforded 5′-substituted phthalimide 168α. Double deprotection of 168α with hydrazine hydrate yielded (−)-157. Alternatively, synthesis of (+)-156 was attained in 62% overall yield from β-anomer 164β by the same synthetic procedure. Their configuration counterparts, (−)-156 and (+)-157 were also synthesized starting from D-glutamic acid by the same methodology.108) This synthetic approach should enable the supply of a variety of derivatives by which their biological activity can be assessed.

Chart 40. Transformation of α-Anomer164α into trans-Isomer (−)-157
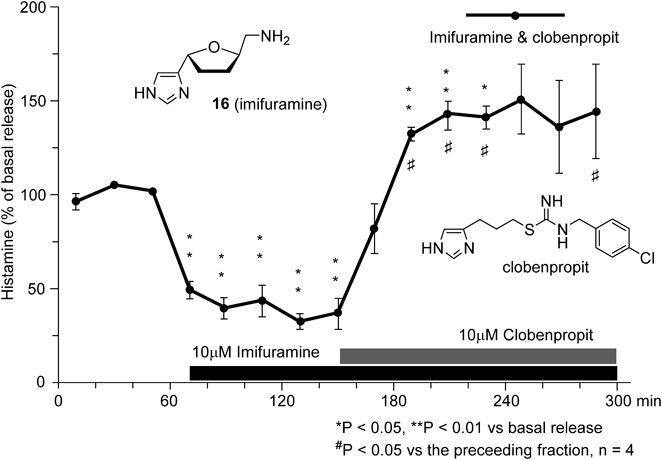
Using an in vivo microdialysis method109) (Fig. 19), we examined whether the four synthesized stereoisomers had any effect on the release of histamine in the brain of rats. Among them, only (+)-157, imifuramine exhibited H3R-agonistic activity.107) Administration of 10 µM imifuramine to the perfusion fluid decreased histamine release to about 30% of the basal release, as shown in Fig. 20. Furthermore, co-infusion of the H3R-antagonist, clobenpropit (10 µM),104) fully antagonized this effect and increased histamine release to about 160% of basal levels107) (Fig. 20). Activity of imifuramine measured by microdialysis was approximately equal to that of immepip.104) These observations support the H3R-agonistic activity of imifuramine. The other three isomers had no effect on histamine release at 10 µM concentration. Therefore, imifuramine was indicated as a novel type of histamine H3R-agonist, suggesting the extended model B as bioactive conformations of the H3-agonists107,108) (Fig. 15).
7.2. Development of the First Selective Human H4-Receptor Agonist OUP16110)A new histamine receptor, H4R was discovered by several groups in the quest for new G-protein-coupled receptors (GPCR).103) The overall amino acid sequence showed approximately 37% homology between H3Rs and H4Rs, as illustrated in Fig. 14. However, the distribution of human H4R (hH4R) mRNA was entirely different from that of the H3R. Although little is known about H4R’s in vivo function, its expression in bone marrow and eosinophils may suggest a potential new role for histamine in regulation of hemopoietic and immune functions (Fig. 14). To investigate the possible physiological function of H4R, a specific ligand is required. Yet, most H3R ligands are active at the H4R as well. For example, the classical selective H3R agonist, (R)-α-methylhistamine shows H4R-agonistic activity, and the H3R-antagonist prototype thioperamide has moderate affinities for H4R. No ligands have been reported that can selectively target H4R.
In the study described above, we synthesized the respective four stereoisomers of the THFs (Fig. 16) and 2′- and 5′-dihydrofurans (DHFs)108) containing imidazole. Furthermore, a series of 16 compounds related to the chiral THFs were synthesized and examined in vivo by radioligand displacement studies and functional assays for H3Rs and H4Rs expressed in SK-N-MC cells.110) Among them, the (2S,5S)-isomer (−)-157 of amino compounds showed approximately 300-fold higher selectivity at the H3R than at the H4R, as indicated in Fig. 21. On the other hand, (2R,5S)- and (2R,5R)-cyanoguanidine derivatives OUP-13 and OUP-16 bound to the H4R with a pEC50 value of 6.7 and 7.1, respectively, and exhibited full agonistic activities (α = 1.0) at the H4R with 46- and 41-fold higher potency than at the H3R, respectively. As such, OUP-13 and OUP-16 are the first selective H4R agonists.110)
In relation to the development of selective H4R ligands, improved synthesis of imifuramine and its derivatives was achieved via cyclization of a diazafulvene intermediate generated by Bu3P/TMAD treatment of a diol intermediate.82)
7.3. Development of Novel H3R Antagonists OUP-133 and 153The (2R,5S)-trans- and (2S,5S)-cis-stereoisomers 174a and 174b of 4(5)-(5-aminotetrahydropyanyl-2-yl)imidazole, which have two chiral centers and adopt a stable chair conformation, were efficiently synthesized via cyclization of diol intermediate 170 using L-glutamine as starting material, as shown in Chart 41.111) Their enantiomers, (2S,5R)-trans- and (2R,5R)-cis-isomers were synthesized by the same methodology from D-glutamine.111) Their stereoisomers were converted into cyanoguanidines, and into N-isopropyl and N-3,3-dimethylbutyl derivatives, respectively.111)

Chart 41. Synthesis of Chiral 4(5)-(5-Aminotetrahydropyanyl-2-yl)imidazoles
The results of in vivo brain microdialysis of the derivatives apparently indicated that (2S,5R)-isomers increased release of neuronal histamine. Among the many (2S,5R)-N-alkyl derivatives, OUP-133 and OUP-153 increased histamine release to 180–190% and 180–200% of basal levels, respectively, and were novel histamine H3 antagonists (Fig. 22).
7.4. Design and Synthesis of Novel Nonimidazole H3R Antagonist OUP-186100)Early imidazole-based H3R antagonists are widely used in pharmacology as potent prototype H3R antagonists such as thioperamide and clobenpropit.102) However, imidazole-derived ligands have an inhibitory effect on CYP enzymes, caused by complexation of the imidazole nitrogen atom to the iron center at the active site of the enzyme, thus leading to drug-drug interactions114) (Fig. 23). Moreover, because imidazole is a strong hydrogen-bond donor and acceptor, some ligands exhibit poor oral bioavailability and difficulty in penetrating the blood brain barrier for entry into the central nervous system115) (Fig. 23).
Recently, a number of new H3R antagonists have been reported and are currently being evaluated in clinical trials.116) These non-imidazole-based antagonists contain a range of different cyclic amines, such as piperidine, homopiperidine, and pyrrolidine. Therefore, the development of potent non-imidazole-based H3R antagonists has become attractive in the search for potential drug candidates.117)
Meanwhile, an extremely potent H3R agonist, clobenpropit is composed of an imidazole ring, a propylspacer and an isothioureas central core that is connected to lipophilic 4-chlorobenzyl group, as indicated in Fig. 23. We designed novel non-imidazole-based H3R antagonists by modification of clobenpropit, substituting the imidazole with typical secondary cyclic amines: pyrrolidine, morphorine, piperidine, methylpiperidine, and piperazine, and containing the different methylene-spacers (n = 1–3)112,113,117) (Fig. 23).
As exemplified in Chart 42, a synthetic method of isothioureas was developed in this investigation. A H3R antagonist target, N-[(4-chlorophenyl)butyl]-S-[3-(piperidin-1-yl)propyl]isothiourea 178 (OUP-186), was synthesized from the starting amine 175 using 2-nitrophenylacetyl isothiocyanate (NPAI) which is a newly designed intercalating agent of SCN atoms.113,117) Reaction of 175 with NPAI afforded thiourea 176 (92%). This thiourea 176 was subsequently converted into S-alkylisothiourea 177 (93%) via Mitsunobu S-alkylation.113,117) Selective elimination of the acetyl moiety of 2-nitrophenylacetyl-protected isothioureas was achieved by catalytic reduction (10% palladium carbon (Pd-C)) of 177 under hydrogen atmosphere.113) The final step proceeded through preferential cleavage of N-CO bond over cleavage of a fissile C–S bond, removing 1-hydroxy-2-oxindole from a hydroxylamine intermediate 179, as shown in Chart 42.113,117) The overall yield of 178 (OUP-186) was therefore 74% over three steps from amine 175.113)

Chart 42. Efficient Synthesis of OUP-186
The synthetic method provided many analogs, in which 178 (OUP-186) showed a potent and selective H3R antagonistic activity (pA2 = 9.6, pIC50 = 8.2) against in vitro hH3R, while being inactive against hH4R (Fig. 24). However, surprisingly. In vivo rat brain microdialysis was completely inactive. This result suggested that 178 was inactive as H3R antagonist in rats, but potent in humans, suggesting differences in antagonist affinities between species. On the other hand, homologue 180 (OUP-181, n = 2), which was tethered to the chlorophenyl group by a C2 chain, gave hH3R antagonistic activity (pA2 = 7.4, pIC50 = 8.1), whereas in vivo rat brain microdialysis increased the histamine release by 140–150% of basal release (Fig. 24).
This discrepancy of 178 between in vitro and in vivo studies prompted us to investigate the structural differences in the ligand-binding cavities of hH3R and rat H3R (rH3R) using molecular modeling.112) Among the amino acids that showed variation between the two species, only Thr119/Ala119 and Ala122/Val122 were found situated in the inner site of transmembrane (TM) region. In silico docking studies of 178 and the C2-shorted homolog 180 with h/rH3Rs were carried out.112) As shown in Fig. 25, three hydrophilic interactions were observed in the 178-hH3R model (Fig. 25A). Similar stable interaction was observed in 180-rH3R model, as shown in Fig. 25B. Because 180 has a more compact structure than 178, it could bind to the shorter cavity of rH3R, filling up by the sidechain of Val122. We next attempted to construct the complex of 178 with rH3R, but failed to produce a stable model, considering the theoretical potential energy of the whole molecular system, because of the van der Waals repulsion between chlorophenylbutyl group of 178 and Val122 side-chain.100) From these findings, it revealed that this species-selectivity of 178 is caused by an Ala122/Val122 mutation between the antagonist-docking cavities in hH3Rs and rH3Rs. In view of the observation that drug development for many human diseases relies heavily on rodent-based models, the discovery that 178 (OUP-186) behaves so differently in the two species is of great significance.
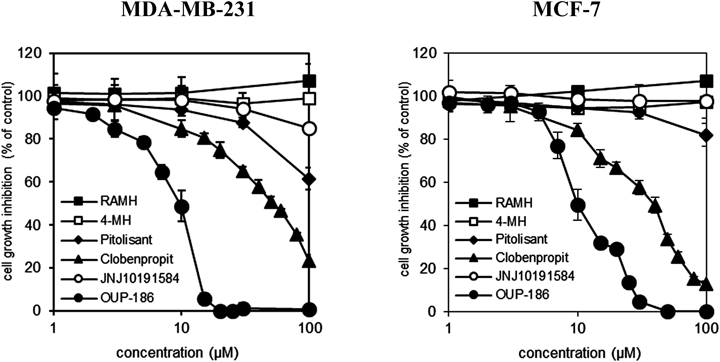
7.6. Human Breast Cancer Cell Proliferation Inhibition of OUP-186118)H3R is expressed in a variety of cancers, suggesting a possible role for histamine in tumorigenesis. H3R expression in breast carcinomas correlates with increases in levels of proliferating cell nuclear antigen and malignancy. Activation of H3R by its agonist induces proliferation of breast carcinoma MDA-MB-231 cells.119) As described above, these data suggest that H3R antagonists may be effective therapeutic agents against breast cancer. OUP-186 exhibited potent and selective H3R antagonistic activity as well as no activity against the hH4R. Therefore, we compared the effects of OUP-186 on proliferation of estrogen receptor negative (ER−) breast cancer cells (MDA-MB-231) and ER+ breast cancer cells (MCF7) versus clobenpropit.118) OUP-186 and clobenpropit suppressed the proliferation of breast cancer cells, as shown in Fig. 26. The IC50 values at 48 h for OUP-186 and clobenpropit were approximately 10 µM and 50 µM, respectively. Furthermore, OUP-186 potently induced cell death by activating caspase-3/7.118) These results indicate that OUP-186 potently suppresses proliferation and induces caspase-dependent apoptotic death in ER+ and ER− breast cancer cells.
Conclusion
1) CPs having a phosphate moiety are useful and versatile intermediates for introduction of a C1 unit in synthesis of medicines and natural products. Alternatively, the generation of alkylidene carbenes from CPs under neutral condition was developed and its usefulness in organic synthesis was demonstrated.
2) A highly stereoselective synthesis of Z- or E-double bonds in 10-membered thiolcarbonates was achieved by [3,3]- sigmatropic rearrangement of 8-membered thionocarbonates which proceeds through the chair-like versus boat-like transition states. The utility of this methodology was demonstrated by unique and stereoselective synthesis of (−)-yellow scale pheromone.
3) β-Stereoselective synthesis of C4-linked imidazole C-nucleosides via diazafulvene intermediates was developed. Furthermore, imidazole C0- or C2-ribonucleoside PAs with a combination of POM and CE groups were synthesized and thereby incorporated the imidazole nucleosides into the specific position of VS and hairpin ribozymes, successfully probing their catalytic mechanism.
4) Tetrahydrofuranylimidazoles using PhSe group were efficiently synthesized and applied to new hH3R agonist, imifuramine, and the first selective hH4R agonist, OUP-16. Furthermore, S-Alkyl-N-alkylisothiourea compounds were synthesized using NPAI to discover novel non-imidazole H3R antagonists. Among the synthesized compounds, OUP-186 exhibited potent and selective antagonism against hH3R but not hH4R, in vitro. Of particular interest, it did not show antagonism for histamine release in rat brain microdialysis in vivo, suggesting species-selective differences in antagonist affinities. Furthermore, in silico docking studies of OUP-186 and its C2-homolog (OUP-181) in human/rat H3Rs indicated that the structural difference of antagonist-docking sites between human and rat H3Rs was attributable to Ala122/Val122 mutation. In addition, OUP-186 exhibited a potent suppressive activity against proliferation of human breast cancer cells.
Acknowledgments
The experiments performed herein were carried out at our laboratory (Department of Pharmaceutical Organic Chemistry) at Osaka University of Pharmaceutical Sciences (OUPS), and I would like to express heartfelt gratitude to the Emeritus Prof. T. Kurihara of OUPS for his support and encouragement. The following collaborators are appreciated for their invaluable efforts: Dr. R. Yoneda, Dr. L. Araki (Minoura), Dr. H. Yoneyama, Dr. M. Fujitake, Prof. Y. Usami, Prof. A. Yamatodani, Dr. T. Hashimoto, Dr. Y. Sakamoto, Dr. Z. Zhao, Prof. D. M. J. Lilley, Dr. S. Tanaka, the late Dr. D. Yamamoto, and the late Prof. Takaoka. A number of co-workers detailed in the references are also acknowledged for their hard work, creativity, and inventions. Our work was financially supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and from the Japan Society for the Promotion of Science, to whom the author’s thanks are due. I thank Prof. T. Kusunose for valuable support in writing English manuscripts. Finally, I would like to express his grateful acknowledgement to Emeritus Professor T. Shioiri at Nagoya City University for his support and encouragement through all studies.
Conflict of Interest
The author declares no conflict of interest
Notes
This review of the author’s work was written by the author upon receiving the 2019 Pharmaceutical Society of Japan Award for Divisional Scientific Contribution.
References
- 1) Harusawa S., Shioiri T., Tetrahedron, 72, 8125–8200 (2016), and references cited therein.
- 2) Harusawa S., Hamada Y., Shioiri T., Synthesis, 1979, 716–718 (1979).
- 3) Harusawa S., Hamada Y., Shioiri T., Tetrahedron Lett., 20, 4663–4666 (1979).
- 4) Harusawa S., Hamada Y., Shioiri T., Heterocycles, 15, 981–984 (1979).
- 5) Harusawa S., Shiori T., Tetrahedron Lett., 23, 447–448 (1982).
- 6) Harusawa S., Shioiri T., J. Synth. Org. Chem. Jpn., 39, 741–753 (1981).
- 7) Harusawa S., “Studies on Novel Cyanation Reactions Using Diethyl Phosphorocyanidate (DEPC),” Ph.D. Dissertation, Nagoya City University, Nagoya, Japan, 1982.
- 8) Gregory R. J. H., Chem. Rev., 99, 3649–3682 (1999).
- 9) North M., Usanov D. L., Young C., Chem. Rev., 108, 5146–5226 (2008).
- 10) Kurihara T., Harusawa S., Yoneda R., J. Synth. Org. Chem. Jpn., 46, 1164–1178 (1988).
- 11) Harusawa S., Yoneda R., Kurihara T., Hamada Y., Shioiri T., Chem. Pharm. Bull., 31, 2932–2935 (1983).
- 12) Harusawa S., Yoneda R., Kurihara T., Hamada Y., Shioiri T., Tetrahedron Lett., 25, 427–428 (1984).
- 13) Yoneyama H., Numata M., Uemura K., Usami Y., Harusawa S., J. Org. Chem., 82, 5538–5556 (2017).
- 14) Kurihara T., Terada T., Harusawa S., Yoneda R., Chem. Pharm. Bull., 35, 4793–4802 (1987).
- 15) Kurihara T., Terada T., Satoda S., Yoneda R., Chem. Pharm. Bull., 34, 2786–2798 (1986).
- 16) Harusawa S., Nakamura S., Yagi S., Kurihara T., Hamada Y., Shioiri T., Synth. Commun., 14, 1365–1371 (1984).
- 17) Kurihara T., Harusawa S., Jpn. Kokai Tokkyo Koho, 1985, JP 60184058 A 19850919.
- 18) Yoneda R., Osaki T., Harusawa S., Kurihara T., J. Chem. Soc., Perkin Trans. 1, 607–610 (1990).
- 19) Yoneda R., Osaki H., Harusawa S., Kurihara T., Chem. Pharm. Bull., 37, 2817–2818 (1989).
- 20) Yoneda R., Harusawa S., Kurihara T., J. Org. Chem., 56, 1827–1832 (1991).
- 21) Yoneda R., Harusawa S., Kurihara T., Tetrahedron Lett., 30, 3681–3684 (1989).
- 22) Oldenziel O. H., van Leusen D., van Leusen A. M., J. Org. Chem., 42, 3114–3118 (1977).
- 23) Harusawa S., Miki M., Yoneda R., Kurihara T., Chem. Pharm. Bull., 33, 2164–2167 (1985).
- 24) Kurihara T., Miki M., Yoneda R., Harusawa S., Chem. Pharm. Bull., 34, 2747–2753 (1986).
- 25) Mita T., Fukuda N., Roca F. X., Kanai M., Shibasaki M., Org. Lett., 9, 259–262 (2007).
- 26) Yoneyama H., Usami Y., Harusawa S., Synthesis, 51, 1791–1794 (2019).
- 27) Harusawa S., Yoneyama H., Heterocycles, 96, 2037–2078 (2018).
- 28) Grainger R. S., Munro K. R., Tetrahedron, 71, 7795–7835 (2015).
- 29) Knorr R., Chem. Rev., 104, 3795–3850 (2004).
- 30) Colvin E. W., Hamill B. J., J. Chem. Soc., Chem. Commun., 5, 151–152 (1973).
- 31) Colvin E. W., Hamill B. J., J. Chem. Soc., Perkin Trans. I, 8, 869–874 (1977).
- 32) Gilbert J. C., Weerasooriya U., J. Org. Chem., 44, 4997–4998 (1979).
- 33) Hari Y., Aoyama T., Shioiri T., “Science of Synthesis Knowledge Updates 2010/4,” ed. by Oestreich M., Stuttgart, Thieme, 2011, pp. 25–68.
- 34) Ohira S., Synth. Commun., 19, 561–564 (1989).
- 35) Müller S., Liepold B., Roth G. J., Bestmann H. J., Synlett, 1996, 521–522 (1996).
- 36) Habrant D., Rauhala V., Koskinen A. M. P., Chem. Soc. Rev., 39, 2007–2017 (2010).
- 37) Yoneyama H., Uemura K., Usami Y., Harusawa S., Tetrahedron, 73, 6109–6117 (2017).
- 38) Wittenberger S. J., Donner B. G., J. Org. Chem., 58, 4139–4141 (1993).
- 39) Miwa K., Aoyama T., Shioiri T., Synlett, 1994, 107–108 (1994).
- 40) Miwa K., Aoyama T., Shioiri T., Synlett, 1994, 109 (1994).
- 41) Miwa K., Aoyama T., Shioiri T., Synlett, 1994, 461–462 (1994).
- 42) Gasparini F., Lingenhöhl K., Stoehr N., Flor P. J., Heinrich M., Vranesic I., Biollaz M., Allgeier H., Heckendorn R., Urwyler S., Varney M. A., Johnson E. C., Hess S. D., Rao S. P., Sacaan A. I., Santori E. M., Veliçelebi G., Kuhn R., Neuropharmacology, 38, 1493–1503 (1999).
- 43) Zheng J.-C., Yun S. Y., Sun C., Lee N.-K., Lee D., J. Org. Chem., 76, 1086–1099 (2011).
- 44) Harusawa S., Yoneyama H., Hisaka F., Fujisue D., Usami Y., Zhao Z., Hetrocycles, 96, 106–126 (2018).
- 45) Cantillo D., Gutmann B., Kappe C. O., J. Am. Chem. Soc., 133, 4465–4475 (2011).
- 46) Ohira S., Ida T., Moritani M., Hasegawa T., J. Chem. Soc., Perkin Trans. 1, 2, 293–298 (1998).
- 47) Ohira S., Ishi S., Shinohara K., Nozaki H., Tetrahedron Lett., 31, 1039–1040 (1990).
- 48) Yoneyama H., Uemura K., Usami Y., Harusawa S., Tetrahedron, 74, 2143–2150 (2018).
- 49) Yaginuma S., Muto N., Tsujino M., Sudate Y., Hayashi M., Otani M., J. Antibiot., 34, 359–366 (1981).
- 50) Herdewijin P., Modified Nucleosides, “Biochemistry, Biotechnology and Medicine,” Wiley-VCH, Weinheim, 2008
- 51) Tsujino M., Yaginuma S., Fujii T., Hayano K., Matsuda T., Watanabe T., Abe J., Curr. Chemother. Infect. Dis., 3, 1559 (1981).
- 52) Chen Q., Davidson A., Bioorg. Med. Chem. Lett., 27, 4436–4439 (2017).
- 53) Ohira S., Sawamoto T., Yamato M., Tetrahedron Lett., 36, 1537–1538 (1995).
- 54) Kim S., Cho C. M., Tetrahedron Lett., 36, 4845–4848 (1995).
- 55) Harusawa S., Kurihara T., “Reviews on Heteroatom Chemistry,” Vol. 16, ed. by Oae S., MYU, Tokyo, 1997, pp. 137–169.
- 56) Holton R. A., Juo R. R., Kim H. B., Williams A. D., Harusawa S., Lowenthal R. E., Yogai S., J. Am. Chem. Soc., 110, 6558–6560 (1988).
- 57) Harusawa S., Tomii S., Takehisa C., Ohishi H., Yoneda R., Kurihara T., Chem. Pharm. Bull., 40, 2279–2282 (1992).
- 58) Harusawa S., Kurokawa T., Fujii H., Yoneda R., Kurihara T., Chem. Pharm. Bull., 37, 2567–2569 (1989).
- 59) Harusawa S., Osaki H., Kurokawa T., Fujii H., Yoneda R., Kurihara T., Chem. Pharm. Bull., 39, 1659–1667 (1991).
- 60) Harusawa S., Osaki H., Fujii H., Yoneda R., Kurihara T., Tetrahedron, 48, 9433–9450 (1992).
- 61) Harusawa S., Osaki H., Fujii H., Yoneda R., Kurihara T., Tetrahedron Lett., 31, 5471–5474 (1990).
- 62) Harusawa S., Osaki H., Yoneda R., Kurihara T., Ohishi H., Tetrahedron Lett., 32, 1203–1206 (1991).
- 63) Harusawa S., Osaki H., Takemura S., Yoneda R., Kurihara T., Tetrahedron Lett., 33, 2543–2546 (1992).
- 64) Harusawa S., Takemura S., Osaki H., Yoneda R., Kurihara T., Tetrahedron, 49, 7657–7666 (1993).
- 65) Harusawa S., Takemura S., Yoneda R., Kurihara T., Tetrahedron, 49, 10577–10586 (1993).
- 66) Harusawa S., Kase N., Yoneda R., Kurihara T., Tetrahedron Lett., 35, 1255–1258 (1994).
- 67) Harusawa S., Moriyama H., Kase N., Ohishi H., Yoneda R., Kurihara T., Tetrahedron, 51, 6475–6494 (1995).
- 68) Harusawa S., Araki L., Kurihara T., J. Synth. Org. Chem. Jpn., 61, 682–693 (2003).
- 69) Araki L., Harusawa S., J. Pharm. Soc. Japan, 130, 1707–1724 (2010).
- 70) Levy D. E., Tang C., “The Chemistry of C-Glycosides,” Pergamon Press, Oxford, 1995.
- 71) Wu Q., Simons C., Synthesis, 2004, 1533–1553 (2004).
- 72) Štambaskỳ J., Hocek M., Kočovskỳ P., Chem. Rev., 109, 6729–6764 (2009).
- 73) Gimmett M. R., “Comprehensive Heterocyclic Chemistry II: Imidazoles,” Vol. 3, ed. by Katritzky A. R., Rees C. W., Scriven E. F., Pergamon Press, Oxford, 1996.
- 74) Revuelta J., Machetti F., Cicchi S., “Modern Heterocyclic Chemistry,” Vol. 2, ed. by Alvarez-Builla J., Vaquero J. J., Barluenga J., Wiley-VCH, Weinheim, 2011, pp. 809–923.
- 75) Harusawa S., Murai Y., Moriyama H., Ohishi H., Yoneda R., Kurihara T., Tetrahedron Lett., 36, 3165–3168 (1995).
- 76) Harusawa S., Murai Y., Moriyama H., Imazu T., Ohishi H., Yoneda R., Kurihara T., J. Org. Chem., 61, 4405–4411 (1996).
- 77) Harusawa S., Moriyama H., Murai Y., Imazu T., Ohishi H., Yoneda R., Kurihara T., Hata H., Sakamoto Y., Chem. Pharm. Bull., 45, 53–61 (1997).
- 78) Kurihara T., Araki L., Harusawa S., Suzuki H., Heterocycles, 53, 1957–1973 (2000).
- 79) Kurihara T., Harusawa S., Ijichi I., Araki L., Heterocycles, 53, 2739–2751 (2000).
- 80) Kurihara T., Araki L., Harusawa S., Ijichi I., Ohishi H., Heterocycles, 55, 889–903 (2001).
- 81) Araki L., Harusawa S., Fukuda M., Kurihara T., Heterocycles, 55, 1907–1917 (2001).
- 82) Harusawa S., Araki L., Terashima H., Kawamura M., Takashima S., Sakamoto Y., Hashimoto T., Yamamoto Y., Yamatodani A., Kurihara T., Chem. Pharm. Bull., 51, 832–837 (2003).
- 83) Harusawa S., Matsuda C., Araki L., Kurihara T., Synthesis, 2006, 793–798 (2006).
- 84) “Ribozymes and RNA Catalysis,” ed. by Lilley D. M. J., Eckstein F., Royal Society of Chemistry, Cambridge, 2008.
- 85) Lafontaine D. A., Wilson T. J., Norman D. G., Lilley D. M. J., J. Mol. Biol., 312, 663–674 (2001).
- 86) Lafontaine D. A., Wilson T. J., Zhao Z., Lilley D. M. J., J. Mol. Biol., 323, 23–34 (2002).
- 87) Araki L., Harusawa S., Yamaguchi M., Yonezawa S., Taniguchi N., Lilley D. M. J., Zhao Z., Kurihara T., Tetrahedron Lett., 45, 2657–2661 (2004).
- 88) Araki L., Harusawa S., Yamaguchi M., Yonezawa S., Taniguchi N., Lilley D. M. J., Zhao Z., Kurihara T., Tetrahedron, 61, 11976–11985 (2005).
- 89) Zhao Z., Mcleod A., Harusawa S., Araki L., Yamaguchi M., Kurihara T., Lilley D. M. J., J. Am. Chem. Soc., 127, 5026–5027 (2005).
- 90) Wilson T. J., Ouellet J., Zhao Z., Harusawa S., Araki L., Kurihara T., Lilley D. M. J., RNA, 12, 980–987 (2006).
- 91) Harusawa S., Fujitake M., Kurihara T., Zhao Z., Lilley D. M. J., “Current Protocols in Nucleic Acid Chemistry,” ed. by Beaucage S. L., Bergstrom D. E., Herdewijin P., Matsuda A., John Wiley & Sons, New York, 2006, pp. 10.11.1–10.11.15.; therein the Basic and Alternate protocols were described in detail.
- 92) Fujitake M., Harusawa S., J. Pharm. Soc. Japan, 133, 823–841 (2013).
- 93) Fujitake M., Harusawa S., Araki L., Yamaguchi M., Lilley D. M. J., Zhao Z., Kurihara T., Tetrahedron, 61, 4689–4699 (2005).
- 94) Fujitake M., Harusawa S., Zhao Z., Lilley D. M. J., Kurihara T., Bull. Osaka Univ. Pharm. Sci., 1, 107–112 (2007).
- 95) Harusawa S., Araki L., Zhao Z., Lilley D. M. J., Heterocycles, 81, 1861–1869 (2010).
- 96) Araki L., Morita K., Yamaguchi M., Zhao Z., Wilson T. J., Lilley D. M. J., Harusawa S., J. Org. Chem., 74, 2350–2356 (2009).
- 97) Harusawa S., Fujii K., Nishiura M., Araki L., Usami Y., Zhao Z., Lilley D. M. J., Heterocycles, 83, 2041–2055 (2011).
- 98) Harusawa S., Yoneyama H., Fujisue D., Nishiura M., Fujitake M., Usami Y., Zhao Z., McPhee S. A., Wilson T. J., Lilley D. M. J., Tetrahedron Lett., 53, 5891–5894 (2012).
- 99) Harusawa S., Yoneyama H., Usami Y., Yamamoto D., Zhao Z., Synthesis, 46, 2815–2825 (2014).
- 100) Yoneyama H., Yamamoto D., Yamatodani A., Harusawa S., J. Pharm. Soc. Japan, 136, 1217–1232 (2016).
- 101) Arrang J. M., Garbarg M., Schwartz J. C., Nature (London), 302, 832–837 (1983).
- 102) Vollinge R. C., de Koning J. P., Jansen F. P., Leurs R., Menge W. M. P. B., Timmerman H., J. Med. Chem., 37, 332–333 (1994).
- 103) Oda T., Morikawa N., Saito Y., Masuho Y., Matsumoto S., J. Biol. Chem., 275, 36781–36786 (2000).
- 104) Van der Goot H., Schepers M. J. P., Sterk G., Timmerman H., Eur. J. Med. Chem., 27, 511–517 (1992).
- 105) Nagy P. I., Durant G. J., Hoss W. P., Smith D. A., J. Am. Chem. Soc., 116, 4898–4909 (1994).
- 106) Mazurek A. P., Karpinska G. Z., Naturforsch, 49, 471–475 (1994).
- 107) Harusawa S., Imazu T., Takashima S., Araki L., Ohishi H., Kurihara T., Yamamoto Y., Yamatodani A., Tetrahedron Lett., 40, 2561–2564 (1999).
- 108) Harusawa S., Imazu T., Takashima S., Araki L., Ohishi H., Kurihara T., Sakamoto Y., Yamamoto Y., Yamatodani A., J. Org. Chem., 64, 8608–8615 (1999).
- 109) Mochizuki T., Yamatodani A., Okakura K., Takemura M., Inagaki N., Wada H., Naunyn Schmiedebergs Arch. Pharmacol., 343, 190–195 (1991).
- 110) Hashimoto T., Harusawa S., Araki L., Zuiderveld O. P., Smit M. J., Imazu T., Takashima S., Yamamoto Y., Sakamoto Y., Kurihara T., Leurs R., Bakker R. A., Yamatodani A., J. Med. Chem., 46, 3162–3165 (2003).
- 111) Harusawa S., Kawamura M., Araki L., Taniguchi R., Yoneyama H., Sakamoto Y., Kaneko N., Nakao Y., Hatano K., Fujita T., Yamamoto R., Kurihara T., Yamatodani A., Chem. Pharm. Bull., 55, 1245–1253 (2007).
- 112) Harusawa S., Sawada K., Magata T., Yoneyama H., Araki L., Usami Y., Hatano K., Yamamoto K., Yamamoto D., Yamatodani A., Bioorg. Med. Chem. Lett., 23, 6415–6420 (2013).
- 113) Yoneyama H., Magata T., Uemura K., Usami Y., Tanaka S., Takaoka M., Harusawa S., Synthesis, 47, 1291–1302 (2015).
- 114) Yang R., Hey J. A., Aslanian R., Rizzo C. A., Pharmacology, 66, 128–135 (2002).
- 115) Haas H., Panula P., Nat. Rev. Neurosci., 4, 121–130 (2003).
- 116) Schwartz J.-C., Br. J. Pharmacol., 163, 713–721 (2011).
- 117) Yoneyama H., Shimoda A., Araki L., Hatano K., Sakamoto Y., Kurihara T., Yamatodani A., Harusawa S., J. Org. Chem., 73, 2096–2104 (2008).
- 118) Tanaka S., Sakaguchi M., Yoneyama H., Usami Y., Harusawa S., Biochem. Biophys. Res. Commun., 480, 479–485 (2016).
- 119) Medina V., Croci M., Crescenti E., Mohamad N., Sanchez-Jiménez F., Massari N., Nuñez M., Cricco G., Martin G., Bergoc R., Rivera E. S., Cancer Biol. Ther., 7, 28–35 (2008).