Abstract
The total syntheses of dimeric indole alkaloids, haplophytine, and T988s are described. These dimeric compounds comprising two structurally different indole units are ubiquitous in nature, and many possess pharmaceutically important activities. To realize an efficient chemical synthesis of these dimeric indole alkaloids, the establishment of convergent synthetic strategies and development of new coupling methods are indispensable. The linkage of two highly functionalized units at a late stage of the synthesis frequently induces synthetic problems such as chemoselectivity and steric repulsion. Moreover, although transition metal-catalyzed reactions are usually an effective method for the cross-coupling of two units, the application of these cross-coupling reactions to bond formation involving a sterically hindered C(sp3) is often difficult. Thus, even with precise modern synthetic methods, it is currently difficult to realize convergent syntheses of dimeric indole alkaloids possessing a quaternary carbon linking two units. To combat these synthetic problems, we developed a synthetic method to link two indole units using an Ag-mediated nucleophilic substitution reaction. In this review, we provide a detailed discussion of convergent synthetic strategies and coupling methods for dimeric indole alkaloids.
1. Introduction
Various dimeric compounds comprising two identical or similar core skeletons have been found in nature.1–4) Greer and colleagues reported that dimeric compounds account for about 17% of all natural products.1) Interestingly, bioactive compounds comprising two similar core skeletons frequently display greater potency and unique biological activities compared with the corresponding monomeric compounds due to their higher affinity for their target receptors or higher stability upon forming a complex with their target receptors.1,2) Moreover, these dimeric structures facilitate the generation of considerable structural diversity within an expanded chemical space. Among these dimeric compounds, dimeric monoterpenoid indole alkaloids,5) typified by vinblastine and vincristine,6) are a pharmaceutically important class of natural products because several compounds in this class exhibit physiologically valuable biological activities (Fig. 1). Notably, the related compound vinflunine was recently approved for the treatment of adult patients with advanced or metastatic transitional cell carcinoma of the urothelial tract.7) In addition to dimeric monoterpene alkaloids, dimeric pyrroloindole alkaloids have also attracted considerable interest from synthetic chemists and biologists5,8–12) (Fig. 1). One noteworthy compound, chaetocin, was identified as the first known inhibitor of lysine-specific histone methyltransferases.13) Therefore, these dimeric compounds, which possess molecular weights that deviate from Lipinski’s rule, are anticipated to be useful as new drug candidates in the middle molecule drug discovery.14)
The efficient chemical synthesis of these dimeric compounds requires the establishment of convergent synthetic strategies and the development of new coupling methods for connecting the two units. Since the mid-20th century, various cross-coupling reactions such as Suzuki coupling,15) Migita–Kosugi–Stille coupling,16,17) Kumada–Tamao–Corriu coupling,18,19) Hiyama coupling,20) and Negishi coupling21) have been extensively developed. These cross-coupling reactions are reliable methods for the formation of the following bonds: C(sp2)–C(sp2), C(sp2)–C(sp), and C(sp2)–or C(sp)–sterically unhindered C(sp3). However, these reactions frequently result in failure for bond formations involving sterically hindered C(sp3), even for simple substrates. Moreover, highly convergent synthetic approaches involving the linkage of highly functionalized monomers in the late stages of the synthesis frequently induce various synthetic problems such as steric repulsion, regioselectivity, stereoselectivity, and chemoselectivity. These problems often present a formidable barrier to synthetic chemists and prevent the realization of the convergent synthesis of dimeric compounds. Thus, although dimeric alkaloids have attracted interest as fascinating target compounds from the synthetic and medicinal community, the lack of an efficient synthetic methodology makes the supply of these compounds in adequate quantities for drug discovery difficult. With this synthetic background, our group undertook synthetic studies of haplophytine, which seems to be one of the most synthetically difficult dimeric indole alkaloids. Herein, I report the first total synthesis of haplophytine22) via a silver salt-mediated coupling reaction between two indole units and its second-generation synthesis23) through a convergent synthetic route. Furthermore, we applied the silver bis(trifluoromethanesulfonyl)imide (AgNTf2)-mediated coupling reaction discovered in the synthesis of haplophytine to the introduction of aryl moiety into pyrroloindoline24) and total syntheses of T988s.25)
2. Total Synthesis of (+)-Haplophytine
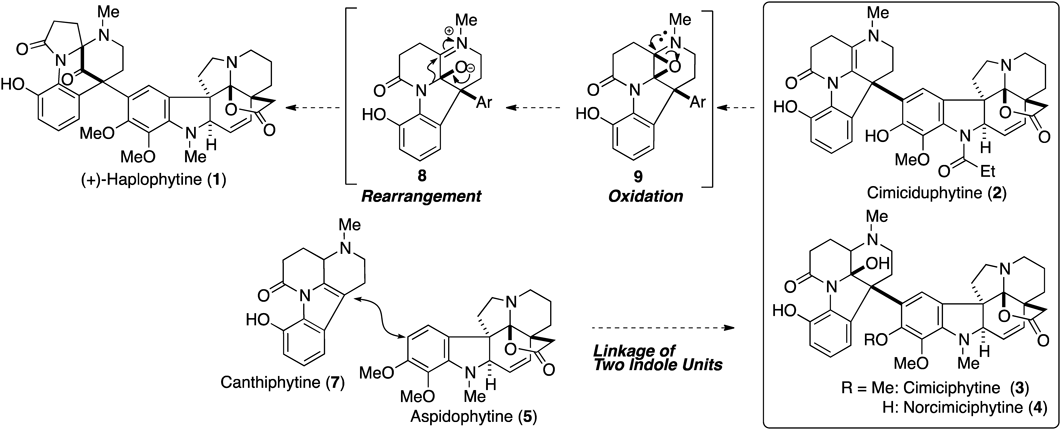
The dimeric monoterpene alkaloid, haplophytine (1), was first isolated from the dried leaves of the plant Haplophyton cimicidum, which has been used as an insecticide against cockroaches and various other insects since Aztec times in parts of Mexico and Central America.26–32) Isolations of various congeners containing cimiciduphytine (2), cimiciphytine (3), and norcimiciphytine (4) from the leaves of the same plant were later reported,33–36) and it was found that these compounds exhibit acetylcholinesterase inhibitory action.37) Compound 1 has attracted considerable attention from numerous synthetic chemists38–44) because of its unique dimeric indole structure containing ten rings and six stereogenic centers. The characteristic structural features of 1 are as follows: i) the left-hand unit comprising a unique diazabicyclo[3.3.1]nonane skeleton including a bridged ketone and aminal functionalities, ii) the right-hand unit comprising a hexacyclic aspidosperma alkaloid, and iii) a quaternary carbon center connecting the two indole units at the bridgehead position. Although much synthetic effort was devoted to the synthesis of 1, its formidable structural complexity precluded successful chemical synthesis until our first total synthesis of 1,22) even though seven total syntheses of (−)-aspidophytine (5) have been reported.45–54) A few months after our report, Nicolaou and Chen’s group also achieved the total synthesis of 1.55)
The most important challenge when designing a synthesis of 1 is linking both units with an accompanying construction of the quaternary carbon center at the bridgehead position. In order to solve the challenging synthetic task involving the formation of the highly fused left-hand framework, we considered the biosynthetic pathway of 1, hoping it might give a hint that could be applied in our synthesis. When considering the biosynthetic pathway, we focused on the structural determination by Cava and Yates’ group.32) Initially, compound 1′ bearing an epoxy group was proposed as the structure of 1 because structure 6, determined by X-ray crystallography, was obtained by treatment of 1 with HBr31) (Fig. 2). After careful spectral analysis, Cava and Yates’ group revised the proposed structure 1′ to the corrected structure 1. The structure of 1 was considered to change to the zwitterion 6 through a unique acid-mediated 1,2-cationic shift. Furthermore, they reported that the natural form of 1 was recovered from the iminium bromide 6 under basic conditions. In addition to the inherent skeletal rearrangement of 1, isolations of the congeners possessing lower oxidation phases relative to 1 suggested the left-hand bicyclo[3.3.1]skeleton of 1 would be formed at the end of the biosynthesis. Based on these reports, the biosynthetic pathway of 1 was proposed to occur, as shown in Fig. 3. Coupling between canthiphytine (7) and 5, which was obtained by an acid-mediated degradation of 1, would afford polycyclic intermediates like 2, 3, and 4. Finally, 1 was proposed to be biosynthesized through the oxidation of the 1,2-diaminoethene moiety and subsequent rearrangement.
According to the proposed biosynthetic pathway of 1, synthetic studies commenced with the construction of the unprecedented left-hand framework by oxidative skeletal rearrangement of a precursor possessing a 1,2-diaminoethene moiety. To carry out the oxidative skeletal rearrangement, two electron-rich aromatic rings must be connected while simultaneously constructing the quaternary carbon center. Position C-4a of tetrahydro-β-carboline, corresponding to position C-3 of the indole, generally acts as a nucleophilic site. Taking advantage of the nucleophilicity at this position, Nicolaou and Chen’s group succeeded in the construction of the quaternary carbon center using an electrophilic substitution reaction of 10 with a combination of catechol derivative 11 and PhI(OCOCF3)242,55) (Fig. 4-a). However, this approach resulted in a low yield of the coupling product and limited the scope for the substrate to a simple indoline. To improve the coupling yield, Chen and colleagues used benzoquinone as an electrophile, but changing the electrophile increased the number of chemical steps required.56) On the other hand, we devised a nucleophilic substitution reaction based on an umpolung strategy, involving generation of a carbocation at the C-4a position that usually acts as a nucleophilic site (Fig. 4-b). Coupling of the two electron-rich aromatic rings was expected to be achieved via the introduction of a leaving group to tetrahydro-β-carboline 16 followed by treatment of 17 with an additive to activate the leaving group in the presence of the right unit 19.
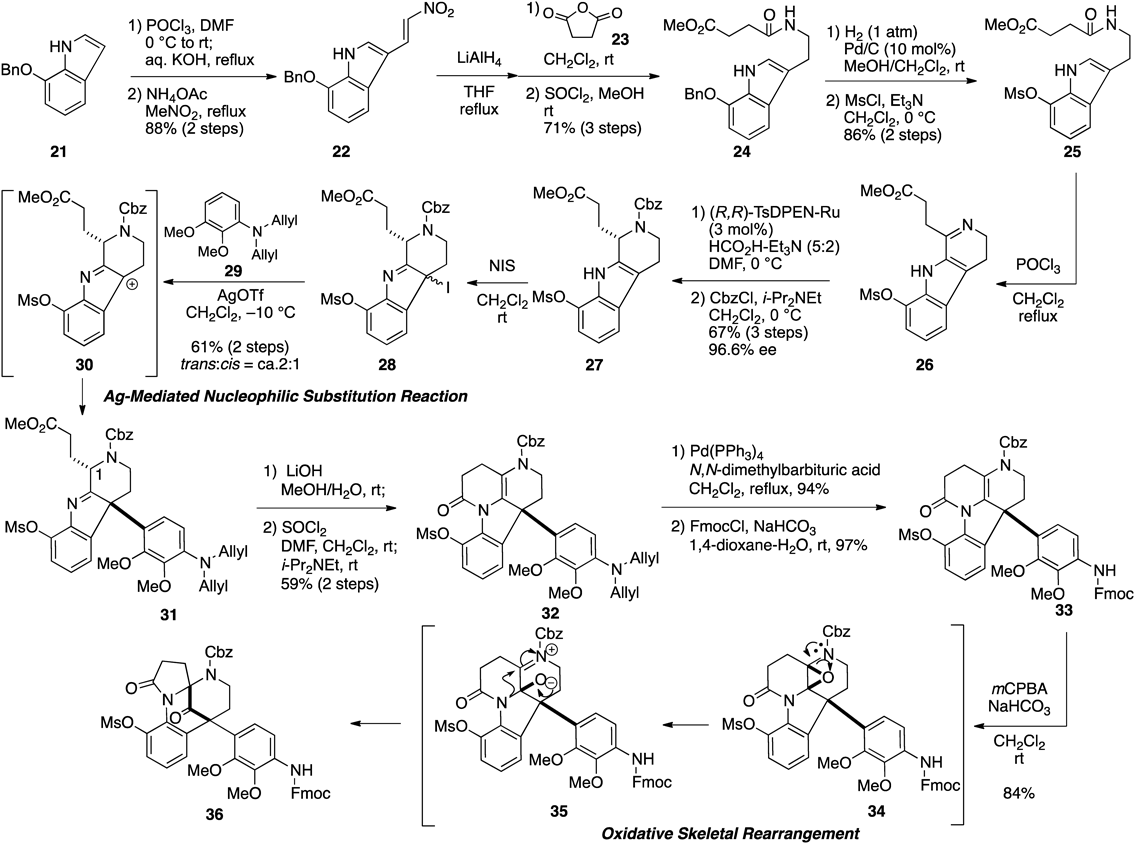
Based on our proposed synthetic strategy, we set out to prepare the tetrahydro-β-carboline in an optically active form (Fig. 5). First, formylation of 7-benzyloxyindole 2122) was conducted under Vilsmeiyer–Haack conditions57) followed by the addition of nitromethane to afford nitroalkene 22. Reduction of nitroalkene followed by acylation with succinic anhydride gave the tryptamide, which was converted to methyl ester 24 with SOCl2 in MeOH. After replacing the benzyl protection with mesyl protection, cyclization of the mesylate under Bischler–Napieralski conditions58) provided dihydro-β-carboline 26. The imine was then subjected to Noyori asymmetric reduction at 0°C,59) followed by protection of the secondary amine with benzyloxycarbonyl chloride (CbzCl) to afford tetrahydro-β-carboline 27 in 67% yield and 96.6% ee after three steps. When the asymmetric reduction was conducted at room temperature, the desired carboline was only obtained in moderate yield due to lactone formation as a side reaction.
With 27 in hand, we undertook the development of the coupling reaction using a nucleophilic substitution strategy. After extensive investigation, the coupling between 27 and an aniline derivative as an alternative to the right-hand unit was achieved via the introduction of an iodo leaving group followed by treatment of the iodoindolenine intermediate 28 with silver trifluoromethanesulfonate (AgOTf) in the presence of 2,3-dimethoxyl aniline 29.22,41) In this coupling reaction, trans-31 was obtained preferentially, presumably due to steric repulsion between 29 and the side chain at the C-1 position. Fortunately, a mixture of diastereomers of 31 was readily separated using a silica chromatography column.
Having successfully established the coupling method, we then focused on the construction of the framework of the left-hand unit according to the proposed oxidative skeletal rearrangement. After the formation of the lactam ring and replacement of two allyl groups with an 9-fluorenylmethoxycarbonyl (Fmoc) group on the aniline nitrogen, we studied the key step. Upon treatment of substrate 33 with meta-chloroperbenzoic acid (mCPBA), the cascade reaction, which involved epoxidation of the 1,2-diaminoethene moiety and a semi-pinacol-type 1,2-shift of the C–N bond, proceeded to provide the desired product possessing a diazabicyclo[3.3.1]nonane skeleton.
Because construction of the left unit skeleton was successful, a pentacyclic aspidosperma compound was used instead of 2,3-dimethoxyaniline as a nucleophile in the Ag-mediated coupling reaction to achieve the convergent total synthesis of 1. Unfortunately, the coupling reaction of iodoindolenine 37 with 5 or even simple indoline 39 did not result in formation of the coupled product 38 (Fig. 6). At this point, we abandoned temporarily the convergent synthetic strategy involving the direct coupling of both polycyclic units and redesigned the synthetic route. When considering an alternative pathway, we focused on Stork’s aspidosperma synthesis,60,61) in which the aspidosperma skeleton was constructed by Fischer indole synthesis62,63) from a phenyl hydrazine and a tricyclic ketone. Thus, the aniline derivative 36 shown in Fig. 5 was considered not as a mere model substrate but a usable compound in an alternative strategy employing Fischer indole synthesis.
Based on the altered synthetic strategy, the total synthesis of aspidophytine was set as our initial goal to validate the synthetic utility of Fischer indole synthesis with the tricyclic ketone prepared by our inventive synthetic route.22) Heating a mixture of tricylic ketone 40 and 2,3-dimethoxyphenylhydrazine (41) in refluxing benzene gave the corresponding hydrazone, which was subjected to Fischer indole synthesis conditions using acetic acid as the solvent at 100°C to afford the desired indolenine compound 42. Compound 5 was then converted from indolenine 42, according to a previously reported procedure46,47) with a slight optimization (Fig. 7-a).
The final stages of the total synthesis of 1 are illustrated in Fig. 7-b. After the conversion of the aniline to phenylhydrazine, we then studied the Fischer indole synthesis reaction between 45 and 40. In this acidic reaction, we were concerned about whether the left structure, which in the case of 1 was readily rearranged with HBr treatment, remained unchanged under acidic conditions with heating. Despite our worries, condensation between 45 and 40 under acidic conditions proceeded smoothly to give the corresponding phenylhydrazine 46 without decomposition of the left-hand skeleton. Initially, Fischer indole synthesis in refluxing acetic acid based on Stork’s conditions was not successful. As a result of an extensive investigation of suitable acids, the yield of 47 was increased to 47% by using a stronger acid, p-toluenesulfonyl acid (TsOH), along with the generation of indole 48 in 27% yield. After the conversion of the imine 47 to a conjugated imine, the Cbz group was removed using BBr3 in the presence of pentamethylbenzene as a cation scavenger64) to liberate the secondary amine. Then, a one-pot 1,2-reduction of imine 49 and reductive methylation of the two secondary amino groups was carried out to give 50. Finally, basic hydrolysis of the ester and phenolic mesylate was followed by the formation of the lactone ring with potassium ferricyanide40) to furnish 1.22) Despite some twists and turns in the synthetic strategy, the total synthesis of 1 was achieved with an Ag-mediated coupling reaction, oxidative skeletal rearrangement, and the use of a classical Fischer indole synthesis reaction. To our knowledge, there are no previous examples of Fischer indole synthesis using phenylhydrazine derivatives comprising structurally complex molecules. We, therefore, believe that we have demonstrated the potential of Fischer indole synthesis for the synthesis of dimeric or polycyclic indole alkaloids.
3. Second-Generation Synthesis of (+)-Haplophytine with a Convergent Strategy
As described above, our group achieved the total synthesis of 1.22) A few months after our synthesis, Nicolaou et al. also reported the total synthesis of 1.55) However, both syntheses left room for improvement in terms of convergence due to their linear synthetic routes involving the construction of the aspidosperma skeleton of the right unit after the formation of the left unit. On the other hand, in the biosynthetic pathway, the aspidosperma right unit is considered to be combined with the left unit via a convergent synthetic pathway (Fig. 3). Based on the biosynthesis hypothesis, we decided to attempt a second-generation synthesis of 1 by a more convergent synthetic strategy through direct coupling between polycyclic units, which we had once abandoned.
We attempted the coupling reaction again using tetrahydro-β-carboline 28 and the pentacyclic aspidosperma compound 43 (Fig. 8). Using the previous method of adding silver salt to a stirred mixture of 28 and 43, the desired coupled product 51 was not obtained. As a result of screening by-products, the formation of reduced compound 27 and iodinated indoline 52 were observed. We presumed that these by-products should be generated through a direct attack of the pentacyclic indoline 43 to an iodo group before the generation of cationic intermediate 30 via elimination of the iodo group. In order to suppress this side reaction, a new operation was incorporated into the reaction procedure in which 43 was added after the cation was already formed by mixing 28 with a silver salt. As a result of this experimental change, the coupling product 51 was obtained for the first time, albeit in low yield. After extensive examination of silver salts including AgOTs, AgBF4, AgPF6, and AgSbF6, we found that only AgNTf265,66) improved the yield, giving trans-51 in 69% yield with a diastereoselectivity of 2.8 : 1.
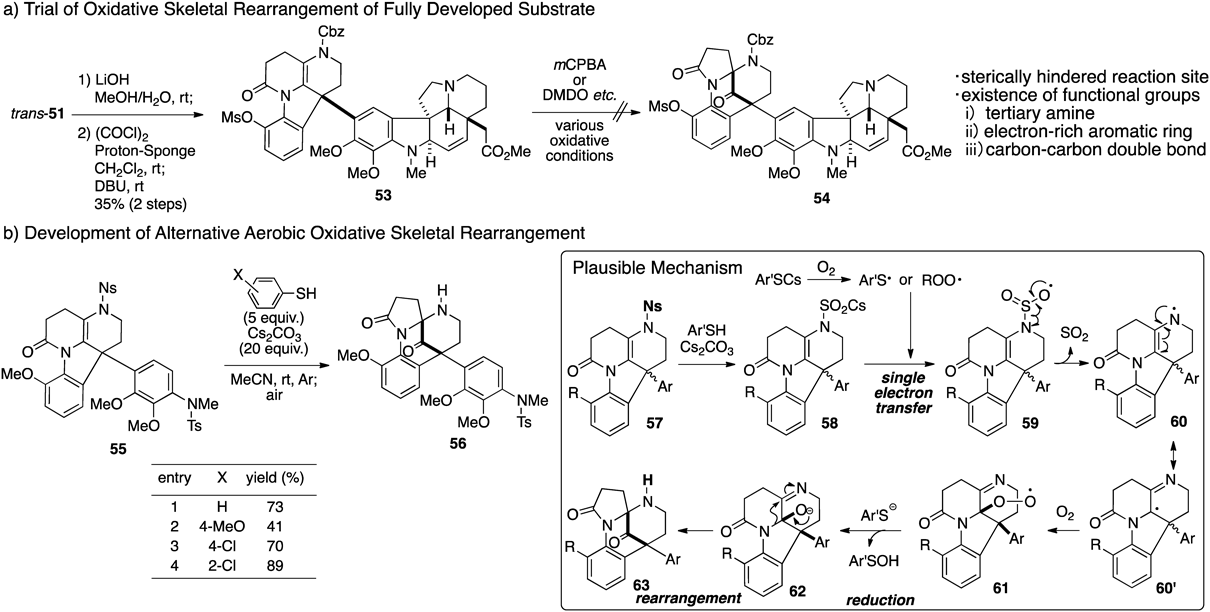
At this point, we expected that the second-generation synthesis of 1 could be readily achieved by the construction of the left-hand skeleton via the oxidative skeletal rearrangement established in the first-generation synthesis. However, a large and unexpected pitfall was encountered; treatment of the fully developed substrate (53) with mCPBA led to decomposition with no formation of the target product 54 (Fig. 9-a). Considering that the reactive site is sterically hindered by the pentacyclic right unit, we tried to use the sterically less hindered dimethyldioxirane (DMDO) as an oxidant. However, the desired product was not obtained at all, and N-oxide formation at the tertiary amine was observed. Although acid was added to the mixture for the purpose of the protonation of the tertiary amine to circumvent the oxidation, this trial also resulted in failure. We faced the synthetic problem of chemoselective oxidation of the substrate 53, which possesses functional groups sensitive to oxidation such as a tertiary amine, an electron-rich aromatic ring, and an alkene. Thus, in order to achieve the biomimetic convergent synthesis of 1, we needed to overcome a synthetic issue related to chemoselectivity. Such issues are often solved with sophisticated enzymatic systems in biosynthetic pathways.
To carry out chemoselective oxidation in the presence of functional groups at a late stage of the synthesis, we planned to increase the relative susceptibility to oxidation of the 1,2-diaminoethene moiety compared with the other functional groups by removal of the protecting group on the nitrogen of the 1,2-diaminoethene. Surprisingly, upon treatment of a model substrate 55 bearing a 2-nitrobenzenesulfonyl (Ns) group with conventional deprotection conditions,67) not only removal of the Ns group but also oxidative skeletal rearrangement proceeded to give the product 56 in 73% yield (Fig. 9-b). Given the unexpected result, we initially considered a reaction mechanism involving deprotection, oxidation, and rearrangement. In this proposed mechanism, the cascade reaction is initiated by removal of the Ns amide to give the anion 58, which after single-electron oxidation by molecular oxygen or thiyl radical species68) generated from thiolate affords 59 with an accompanying extrusion of SO2. The radical intermediate 60 would then immediately trap molecular oxygen to produce peroxyl intermediate 61. Finally, hydroperoxide would be reduced by the thiolate anion, and a subsequent semi-pinacol-type rearrangement of 62 should furnish 63 with a diazabicyclo[3.3.1]nonane skeleton.
Based on our proposed reaction mechanism shown in Fig. 8-b, we examined reaction conditions to elucidate the reaction mechanism and improve the yield. The single-electron oxidation step was determined to be rate determining by tracking the reaction using TLC. To verify the active species involved in the oxidation step, the substitution effect of thiophenols was extensively examined. When the reaction of 55 was conducted using 4-methoxythiophenol, which possesses a lower redox potential than thiophenol,69) the oxidation rate and yield of 56 were decreased. On the other hand, the use of 2-chlorothiophenol, which has a higher redox potential, suppressed the generation of unoxidized products and led to an improvement in the yield of oxidized 56. These control experiments clearly demonstrated the substituent effect of thiophenol in the oxidation step. However, the possibility of molecular oxygen being involved in the oxidation step was not excluded completely. In this way, we discovered an alternative method utilizing molecular oxygen as an oxygen atom source that might solve the synthetic problem of chemoselective oxidation at a late stage of the synthesis.
Having established the aerobic oxidation–skeletal rearrangement cascade, we aimed for a convergent total synthesis of 1 by applying the new oxidation route to a fully elaborated substrate bearing an Ns group (Fig. 10). First, iodoindolenine 64 synthesized from the corresponding tetrahydro-β-carboline was connected to the right unit 65 in the presence of AgNTf2 to give 66 in a total yield of 57% (dr = 2.4 : 1). It should be noted that, in contrast to the Cbz-protected substrate 28, the Ns-protected substrate 64 preferentially afforded cis-66. A computational analysis of the conformation of 64 revealed that the benzene ring of the Ns moiety covered the α-face of the reaction site. Thus, the stereoselectivity might be attributed to steric repulsion between nucleophile 65 and the Ns moiety. After chemoselective saponification70) and lactam formation, trials of conventional Pd-catalyzed deallylation conditions in the presence of 1,3-dimethylbarbituric acid71) or Et3N resulted in failure. After extensive investigation of deallylation conditions, we found the reaction required acid, with pyridinium p-toluenesulfonate (PPTS) being the most effective, to give the deallylated product in high yield. After protection of the indoline with a t-butoxycarbonyl (Boc) group, the substrate 68 was subjected to the slightly refined optimal conditions with 2-chlorothiophenol in the presence of Cs2CO3 in tetrahydrofuran (THF) solvent. As expected, the cascade reaction, including the oxidation step, proceeded smoothly with other functional groups untouched to furnish the desired rearranged product 69 with a high yield of 70%. Removal of the Boc group and reductive methylation of the two secondary amino groups afforded the dimethylated product. Finally, one-pot removal of the tosyl group and saponification of the methyl ester, followed by the oxidative formation of the lactone ring40) furnished 1. Thus, we accomplished the total synthesis of 1 according to a convergent synthetic strategy inspired by the presumed biosynthetic pathway. Achievement of this synthetic project demonstrated that the AgNTf2-mediated nucleophilic substitution reaction was effective for linking two highly functionalized indole units accompanied by the formation of a C(sp2)–C(sp3) bond at a sterically hindered position.
4. Total Synthesis of T988s
Numerous pyrroloindole alkaloids derived from tryptophan are ubiquitous in nature.5,8–12) These pyrroloindole alkaloids possess a wide structural diversity arising from amino acids and a variety of bonding modes containing synthetically difficult quaternary or tetrasubstituted carbon atoms at the sterically hindered C-3a position. Among these compounds, we focused on dimeric indole alkaloids comprising two indole nuclei connected at the C-3a position and planned to apply the Ag-mediated nucleophilic substitution reaction established in the synthetic studies of 1 to pyrroloindole chemistry.
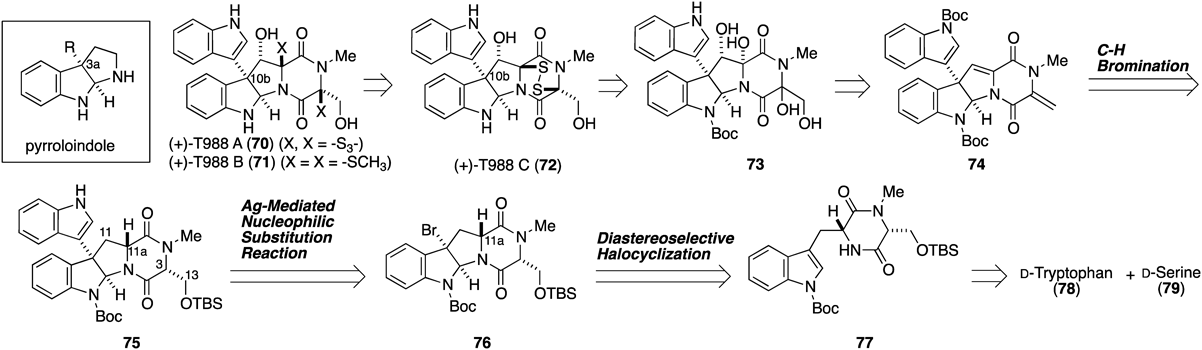
T988 A–C (70–72) equipped with a dioxopiperazine skeleton connected to a pyrroloindole skeleton including a quaternary carbon center with an indolyl group at the C-10b position were isolated from the New Zealand fungi Tilachlidium sp. (CANU-T988) by Munro and and colleagues in 2004.72) T988s are known to possess potent cytotoxicity against cultured P388 leukemia cells. In addition to their fascinating biological activity, the intriguing structures of T988s and their related compounds have inspired a number of synthetic chemists to launch the development of new synthetic methodologies.73–78) Herein, I report our highly efficient total syntheses of T988 A–C (70–72) via AgNTf2-mediated nucleophilic substitution reaction of bromopyrroloindoline.
Our retrosynthetic analysis of 70–72 is illustrated in Fig. 11. The target compounds 70–72 could be synthesized by the construction of the disulfide bridge through bis-1,2-diol intermediate 73. We envisioned that the crucial construction of the C-11–C-11a double bond would be implemented by a sequential C–H bromination at C-11a followed by dehydrobromination. The C-3–C-13 double bond should be easily formed by mild dehydration of a pre-installed hydroxymethyl group using serine. Introduction of a C′3-indole at the C-10b position would be realized by an AgNTf2-mediated nucleophilic substitution reaction of bromopyrroloindoline 76. The bromopyrroloindoline 76 could be readily prepared from D-tryptophan (78) and D-serine (79) according to the procedure reported by Sodeoka and colleagues.79,80)
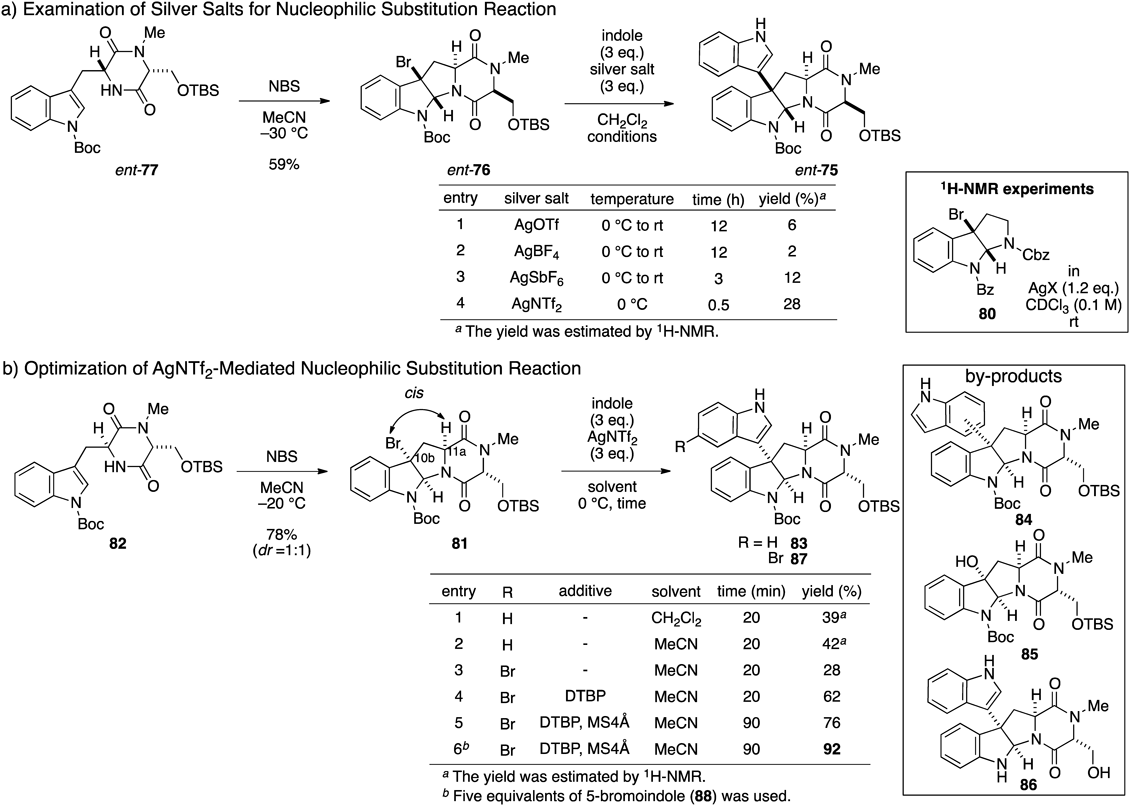
Initially, this project started from the investigation of the AgNTf2-mediated nucleophilic substitution reaction of ent-bromopyrroloindoline 76, which can easily be prepared from natural amino acids. After diastereoselective bromocyclization of ent-77 according to Sodeoka’s procedure, we examined the nucleophilic substitution reaction of ent-76 with various silver salts (Fig. 12-a). In this reaction, a significant difference in reaction rate was observed between AgNTf2 and other silver salts (AgOTf, AgBF4, and AgSbF6). Thus, AgNTf2 gave the best result, as was also found during the synthesis of 1, albeit with an unsatisfactory yield of 76. Furthermore, we performed NMR experiments to compare the halogen activation ability between AgNTf2 and AgOTf. Time-dependent 1H-NMR spectra were measured after the addition of 1.2 equivalents of silver salt (AgNTf2 or AgOTf) to a CDCl3 solution (0.1 M) of bromopyrroloindoline 80 in the absence of a nucleophile. In the case of AgNTf2, the proton signal of 80 disappeared after approximately 20 min. On the other hand, in the case of AgOTf, complete consumption of 80 required about one day. These time-dependent 1H-NMR experiments indicated that the advantage of AgNTf2 over AgOTf can be attributed to its high efficiency in activating the bromo group of bromopyrroloindoline for the generation of the corresponding cationic intermediate, which facilitates conducting the reaction at a lower temperature.
Next, we undertook optimization to improve the product yield in the coupling reaction, which commenced with trials to reduce the steric repulsion between the nucleophile and the reactive site, which is crowded by the dioxopiperazine ring (Fig. 12-b). We expected that the use of the diastereomer (81) of 76 possessing a C-10b–C-11a cis relationship would reduce the steric repulsion between the substrate and the nucleophile to facilitate the coupling reaction. Although this new strategy requires the inversion of the C-11a stereochemistry, this does not affect the overall strategy (Fig. 11) because of the disappearance of the stereochemistry at a later stage. Thus, bromopyrroloindoline 81 was synthesized via bromocyclization of 82 starting from L-tryptophan (ent-78) and D-serine (79). With bromopyrroloindoline 81 having the expected stereochemistry in hand, we conducted the AgNTf2-mediated nucleophilic substitution reaction with the indole. As expected, the yield of the coupled product 83 was improved. Next, we began optimization of the reaction conditions using the substrate 81. Performing the reaction in MeCN solvent instead of CH2Cl2 increased the yield of 83 slightly. As a result of screening by-products, the formation of regioisomer 84, water adduct 85, and deprotected product 86 were observed. Finally, the yield of the desired coupling product 87 was improved to 92% on a gram-scale reaction by addition of appropriate additives such as 2,6-di-tert-butylpyridine and 4 Å molecular sieves in the presence of 5 equivalents of 5-bromoindole (88). Moreover, the established AgNTf2-mediated substitution reaction of bromopyrroloindole proved to be effective for various aromatic compounds, including heterocycles.81,82)
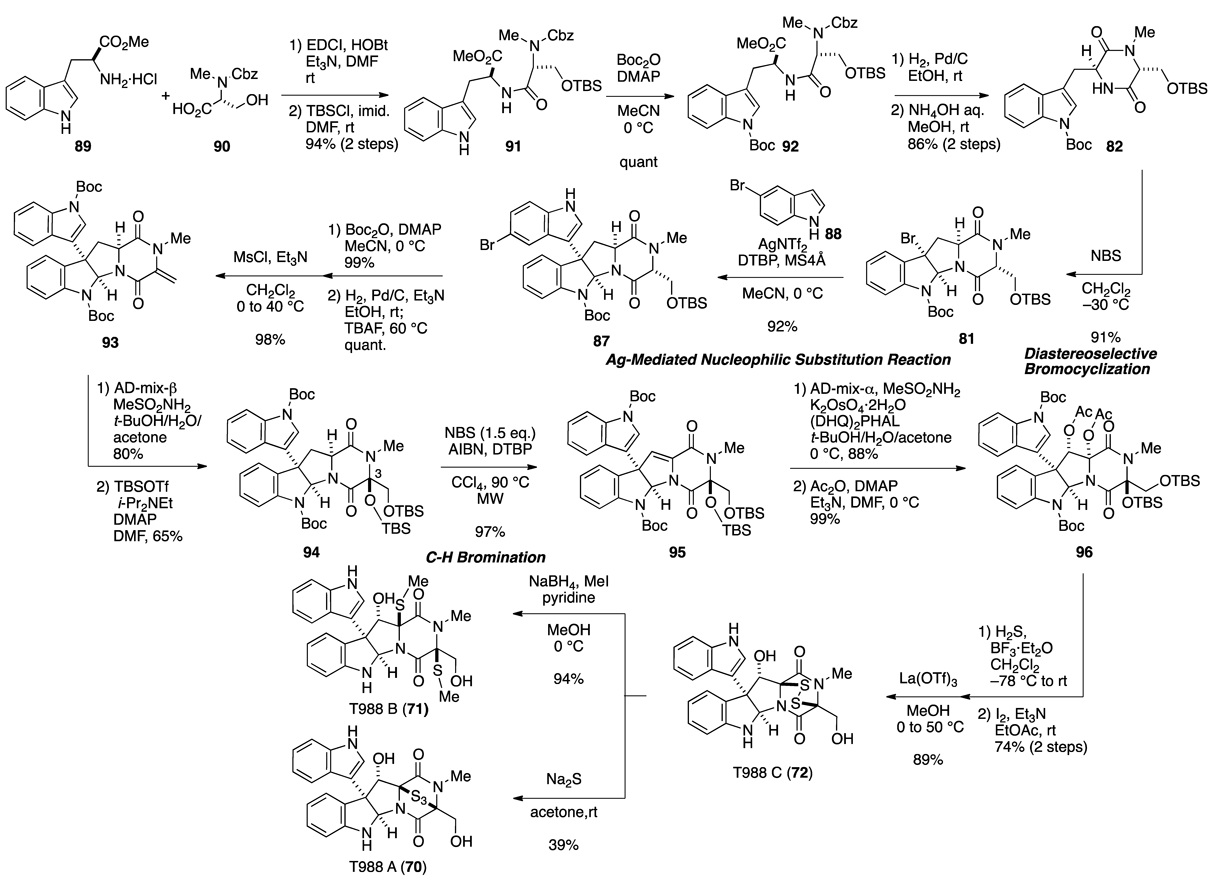
Having established an efficient method for introducing an indolyl group to a pyrroloindole nucleus using AgNTf2-mediated nucleophilic substitution, we started synthetic studies aimed at the total synthesis of T988 A–C (70–72). The synthesis commenced with the condensation of L-tryptophan methyl ester hydrochloride (89) and D-serine derivative 90 (Fig. 13). After sequential protection of the resulting primary alcohol and indole NH, 91 was converted to dioxopiperazine 82 via removal of the Cbz group followed by base treatment. Since bromocyclization of 82 using NBS in MeCN according to Sodeoka’s procedure79,80) gave an approx. 1 : 1 mixture of the desired bromopyrroloindoline 81 and its diastereomer (Fig. 12-b), we extensively investigated the stereoselective bromocyclization conditions. We found that the judicious choice of solvent proved to be crucial for high stereoselectivity. Finally, non-polar solvents such as CH2Cl2 instead of MeCN preferentially yielded the desired diastereomer product 81. Treatment of 81 with 5-bromoindole (88) under optimal conditions furnished 87. Next, we focused on the construction of the dithiodioxopiperazine ring, which was the last remaining task to accomplish the total synthesis of T988s. Initially, protection of the indole NH with a Boc group followed by reductive removal of the Br group on the indole ring and one-pot desilylation gave a primary alcohol. Mesylation of the hydroxy group involved an elimination reaction to provide exo-methylene compound 93. Although the stereochemistry of the C-3 position would disappear at a later stage, diastereoselective dihydroxylation was conducted using AD-mix-β83) for more efficient C–H bromination in a later stage. After protection of the 1,2-diol as a bis-t-butyldimethylsilyl (TBS) ether, the internal alkene at C-11–C-11a was successfully introduced via C–H bromination of 94 at the C-11a position by treatment under bromination conditions with 2,2′-azobisisobutyronitrile (AIBN) and N-bromosuccinimide (NBS) in the presence of 2,6-di-tert-butylpyridine (DTBP) with microwave-assisted heating.84) The internal olefin was subjected to face-selective dihydroxylation of 95 with AD-mix-α to give the expected diol, which was transformed to diacetate 96. The reaction of 96 with hydrogen sulfide in the presence of BF3·OEt2 promoted the removal of TBS and two Boc groups, as well as the formation of dithiol via an N-acyliminium ion according to Overman’s protocol.75) Subsequent formation of disulfide with iodine provided the epidithiodioxopiperazine derivative. Finally, methanolysis with La(OTf)3 in MeOH solvent furnished (+)-T988 C (72). Furthermore, treatment of 72 with NaBH4 in the presence of methyl iodide afforded (+)-T988 B (71), and exposure of 72 to sodium sulfide provided (+)-T988 A (70). Therefore, we accomplished the total synthesis of T988 A–C (70–72). As a notable feature of the synthesis, the overall yield of (+)-T988 C (72) was as high as 18.8%, starting from commercially available L-tryptophan methyl ester hydrochloride (89). These highly efficient total syntheses relied on the development of stereoselective bromocyclization, an AgNTf2-mediated nucleophilic substitution reaction of bromopyrroloindoline with indole, and appropriate stereocontrol of the C-3 position.
Conclusion
This review presented an overview of efficient convergent syntheses of dimeric indole alkaloids. The key to the synthesis of dimeric compounds is the development of synthetic methodologies for linking the two units. In particular, the realization of convergent synthesis requires coupling of both fully elaborated units at a late stage of the synthesis, which entails the following crucial synthetic issues: i) chemoselectivity in the presence of various functional groups and ii) steric factors. Among the many possible dimeric indole alkaloids, we chose haplophytine, which possesses a single bond between two indole nuclei, as a synthetic target, and we undertook the necessary synthetic studies to establish a new synthetic strategy. Through these synthetic studies, we developed new coupling reactions that allow for convergent integration of dimeric indoles via nucleophilic substitution reactions involving cation formation from the haloalkane at the indole C-3 position using silver salts. Above all, these synthetic studies demonstrated that AgNTf2 possesses a high activity for halogen activation compared with other silver salts, and additionally has high chemoselectivity in the presence of nucleophilic heteroatoms and acid-sensitive functionalities. Moreover, the total synthesis of T988s further validates the utility of this method, which enables concise derivatization of the pyrroloindole core. This method is expected to be applied to the synthesis of various dimeric indoles, not limited to the natural products mentioned in this review. We believe that further development of synthetic methodologies for dimeric compounds will provide novel chemical tools with enormous benefits to drug discovery.
This review of the author’s work was written by the author upon receiving the 2018 Pharmaceutical Society of Japan Award for Young Scientists.
Acknowledgments
First, I am grateful to Professor Hidetoshi Tokuyama of Tohoku University for valuable suggestions, heartwarming support, and encouragement in achieving the results described in this review. Since the synthetic project of haplophytine started from Fukuyama laboratory, I would like to thank Emeritus Professor Tohru Fukuyama of The University of Tokyo. The experimental results reviewed in this paper were realized by the dedication and untiring efforts of my collaborators, including Dr. K. Matsumoto, Dr. S. Sumi, Dr. H. Satoh, Dr. K. Ojima, Dr. S. Sato, and Ms. A. Hirayama. The work described herein was supported by a Grant-in-Aid for Research Activity start-up (22890013), Young Scientists (B) (2479003, 26860004), Scientific Research (C) (17K08204) and an Emergency Fund from Astellas Foundation for Research on Metabolic Disorders.
Conflict of Interest
The author declares no conflict of interest.
References
- 1) Voloshchuk T., Farina N. S., Wauchope O. R., Kiprowska M., Haberfield P., Greer A., J. Nat. Prod., 67, 1141–1146 (2004).
- 2) Bérudbé G., Curr. Med. Chem., 13, 131–154 (2006).
- 3) Vrettou M., Gray A. A., Brewer A. R. E., Barrett A. G. M., Tetrahedron, 63, 1487–1536 (2007).
- 4) Lian G., Yu B., Chem. Biodivers., 7, 2660–2691 (2010).
- 5) Kam T.-S., Choo Y.-M., The Alkaloids, 63, 181–337 (2006).
- 6) Haque A., Rahman M. A., Faizi M. S. H., Khan M. S., Curr. Med. Chem., 25, 1650–1662 (2018).
- 7) Gerullis H., Wawroschek F., Köhne C.-H., Ecke T. H., Ther. Adv. Urol., 9, 28–35 (2017).
- 8) Crich D., Banerjee A., Acc. Chem. Res., 40, 151–161 (2007).
- 9) Steven A., Overman L. E., Angew. Chem. Int. Ed., 46, 5488–5508 (2007).
- 10) Schmidt M. A., Movassaghi M., Synlett, 3, 313–324 (2008).
- 11) Ruiz-Sanchis P., Savina S. A., Albericio F., Álvarez M., Chem. Eur. J., 17, 1388–1408 (2011).
- 12) Welch T. R., Williams R. M., Nat. Prod. Rep., 31, 1376–1404 (2014).
- 13) Greiner D., Bonaldi T., Eskeland R., Roemer E., Imhof A., Nat. Chem. Biol., 1, 143–145 (2005).
- 14) Terrett N., Med. Chem. Comm., 4, 474–475 (2013).
- 15) Miyaura N., Yamada K., Suzuki A., Tetrahedron Lett., 20, 3437–3440 (1979).
- 16) Kosugi M., Sasazawa K., Shimizu Y., Migita T., Chem. Lett., 6, 301–302 (1977).
- 17) Milstein D., Stille J. K., J. Am. Chem. Soc., 100, 3636–3638 (1978).
- 18) Tamao K., Sumitani K., Kumada M., J. Am. Chem. Soc., 94, 4374–4376 (1972).
- 19) Corriu R. J. P., Masse J. P., J. Chem. Soc., Chem. Commun., 3, 144a (1972).
- 20) Hatanaka Y., Hiyama T., J. Org. Chem., 53, 918–920 (1988).
- 21) King A. O., Okukado N., Negishi E., J. Chem. Soc., Chem. Commun., 19, 683–684 (1977).
- 22) Ueda H., Satoh H., Matsumoto K., Sugimoto K., Fukuyama T., Tokuyama H., Angew. Chem. Int. Ed., 48, 7600–7603 (2009).
- 23) Satoh H., Ojima K., Ueda H., Tokuyama H., Angew. Chem. Int. Ed., 55, 15157–15161 (2016).
- 24) Tokuyama H., Sato S., Hirayama A., Adachi T., Kawauchi D., Ueda H., Heterocycles, 94, 1940–1957 (2017).
- 25) Sato S., Hirayama A., Ueda H., Tokuyama H., Asian J. Org. Chem., 6, 54–58 (2017).
- 26) Rogers E. F., Snyder H. R., Fischer R. F., J. Am. Chem. Soc., 74, 1987–1989 (1952).
- 27) Snyder H. R., Fischer R. F., Walker J. F., Els H. E., Nussberger G. A., J. Am. Chem. Soc., 76, 2819–2825 (1954).
- 28) Snyder H. R., Fischer R. F., Walker J. F., Els H. E., Nussberger G. A., J. Am. Chem. Soc., 76, 4601–4605 (1954).
- 29) Snyder H. R., Strohmayer H. F., Mooney R. A., J. Am. Chem. Soc., 80, 3708–3710 (1958).
- 30) Saxton J. E., Alkaloids, 8, 673–678 (1965).
- 31) Rae I. D., Rosenberger M., Snabo A. G., Willis C. R., Yates P., Zacharias D. E., Jeffrey G. A., Douglas B., Kirkpatrick J. L., Weisbach J., J. Am. Chem. Soc., 89, 3061–3062 (1967).
- 32) Yates P., MacLachlan F. N., Rae I. D., Rosenberger M., Szabo A. G., Willis C. R., Cava M. P., Behforouz M., Lakshmikantham M. V., Zeiger W., J. Am. Chem. Soc., 95, 7842–7850 (1973).
- 33) Laksmikantham M., Mitchell M. J., Cava M. P., Heterocycles, 9, 1009–1013 (1978).
- 34) Adesomoju A. A., Lakshmikantham M. V., Cava M. P., Heterocycles, 20, 1511–1517 (1983).
- 35) Adesomoju A. A., Rawal V. H., Lakshmikantham M. V., Cava M. P., J. Org. Chem., 48, 3015–3017 (1983).
- 36) Adesomoju A. A., Lakshmikantham M. V., Cava M. P., Heterocycles, 32, 1461–1462 (1991).
- 37) Mroue M. A., Euler K. L., Ghuman M. A., Alam M., J. Nat. Prod., 59, 890–893 (1996).
- 38) Yates P., Schwartz D. A., Can. J. Chem., 61, 509–518 (1983).
- 39) Schwartz D. A., Yates P., Can. J. Chem., 61, 1126–1131 (1983).
- 40) Rege P. D., Tian Y., Corey E. J., Org. Lett., 8, 3117–3120 (2006).
- 41) Matsumoto K., Tokuyama H., Fukuyama T., Synlett, 20, 3137–3140 (2007).
- 42) Nicolaou K. C., Majumder U., Roche S. P., Chen D. Y.-K., Angew. Chem. Int. Ed., 46, 4715–4718 (2007).
- 43) Chughtai M., Eagan J. M., Padwa A., Synlett, 2, 215–218 (2011).
- 44) Dlugosch M., Banwell M. G., Aust. J. Chem., 71, 573–578 (2018).
- 45) He F., Bo Y. X., Altom J. D., Corey E. J., J. Am. Chem. Soc., 121, 6771–6772 (1999).
- 46) Sumi S., Matsumoto K., Tokuyama H., Fukuyama T., Org. Lett., 5, 1891–1893 (2003).
- 47) Sumi S., Matsumoto K., Tokuyama H., Fukuyama T., Tetrahedron, 59, 8571–8587 (2003).
- 48) Mejia-Oneto J. M., Padwa A., Org. Lett., 8, 3275–3278 (2006).
- 49) Marino J. P., Cao G., Tetrahedron Lett., 47, 7711–7713 (2006).
- 50) Mejia-Oneto J. M., Padwa A., Helv. Chim. Acta, 91, 285–302 (2008).
- 51) Nicolaou K. C., Dalby S. M., Majumder U., J. Am. Chem. Soc., 130, 14942–14943 (2008).
- 52) Satoh H., Ueda H., Tokuyama H., Tetrahedron, 69, 89–95 (2013).
- 53) Yang R., Qiu F. G., Angew. Chem. Int. Ed., 52, 6015–6018 (2013).
- 54) Türkmen Y. E., Gravel M., Rawal V. H., J. Org. Chem., 81, 10454–10462 (2016).
- 55) Nicolaou K. C., Dalby S. M., Li S., Suzuki T., Chen D. Y.-K., Angew. Chem. Int. Ed., 48, 7616–7620 (2009).
- 56) Tian W., Chennamaneni L. R., Suzuki T., Chen D. Y.-K., Eur. J. Org. Chem., 2011, 1027–1031 (2011).
- 57) Vilsmeier A., Haack A., Ber, 60, 119–122 (1927).
- 58) Santos L. S., Pilli R. A., Rawal V. H., J. Org. Chem., 69, 1283–1289 (2004).
- 59) Uematsu N., Fujii A., Hashiguchi S., Ikariya T., Noyori R., J. Am. Chem. Soc., 118, 4916–4917 (1996).
- 60) Stork G., Dolfini J. E., J. Am. Chem. Soc., 85, 2872–2873 (1963).
- 61) Lawton G., Saxton J. E., Smith A. J., Tetrahedron, 33, 1641–1653 (1977).
- 62) Fischer E., Jourdan F., Ber., 16, 2241–2245 (1883).
- 63) Robinson B., Chem. Rev., 69, 227–250 (1969).
- 64) Okano K., Okuyama K., Fukuyama T., Tokuyama H., Synlett, 13, 1977–1980 (2008).
- 65) Vij A., Zheng Y. Y., Kirchmeier R. L., Shreeve J. M., Inorg. Chem., 33, 3281–3288 (1994).
- 66) Stricker M., Oelkers B., Rosenau C. P., Sundermeyer J., Chem. Eur. J., 19, 1042–1057 (2013).
- 67) Kan T., Fukuyama T., Chem. Commun., 4, 353–359 (2004).
- 68) Dénès F., Pichowicz M., Povie G., Renaud P., Chem. Rev., 114, 2587–2693 (2014).
- 69) Schmidt am Busch M., Knapp E.-W., J. Am. Chem. Soc., 127, 15730–15737 (2005).
- 70) Nicolaou K. C., Estrada A. A., Zak M., Lee S. H., Safina B. S., Angew. Chem. Int. Ed., 44, 1378–1382 (2005).
- 71) Garro-Helion F., Merzouk A., Guibe F., J. Org. Chem., 58, 6109–6113 (1993).
- 72) Feng Y., Blunt J. W., Cole A. L. J., Munro M. H. G., J. Nat. Prod., 67, 2090–2092 (2004).
- 73) DeLorbe J. E., Jabri S. Y., Mennen S. M., Overman L. E., Zhang F.-L., J. Am. Chem. Soc., 133, 6549–6552 (2011).
- 74) Boyer N., Movassaghi M., Chem. Sci., 3, 1798–1803 (2012).
- 75) DeLorbe J. E., Horne D., Jove R., Mennen S. M., Nam S., Zhang F.-L., Overman L. E., J. Am. Chem. Soc., 135, 4117–4128 (2013).
- 76) Coste A., Kim J., Adams T. C., Movassaghi M., Chem. Sci., 4, 3191–3197 (2013).
- 77) Adams T. C., Payette J. N., Cheah J. H., Movassaghi M., Org. Lett., 17, 4268–4271 (2015).
- 78) Wang L., Jia X., Lei H., Xu Z., Ye T., Synlett, 29, 613–616 (2018).
- 79) Iwasa E., Hamashima Y., Fujishiro S., Higuchi E., Ito A., Yoshida M., Sodeoka M., J. Am. Chem. Soc., 132, 4078–4079 (2010).
- 80) Iwasa E., Hamashima Y., Fujishiro S., Hashizume D., Sodeoka M., Tetrahedron, 67, 6587–6599 (2011).
- 81) Sato S., Hirayama A., Adachi T., Kawauchi D., Ueda H., Tokuyama H., Heterocycles, 94, 1940–1957 (2017).
- 82) Hakamata H., Ueda H., Tokuyama H., Org. Lett., 21, 4205–4209 (2019).
- 83) Kolb H. C., VanNieuwenhze M. S., Sharpless K. B., Chem. Rev., 94, 2483–2547 (1994).
- 84) Caballero E., Avendaño C., Menéndez J. C., Tetrahedron, 55, 14185–14198 (1999).