Abstract
Discovery of novel multifunctional inhibitors targeting acetylcholinesterase (AChE) has becoming a hot spot in anti-Alzheimer’s disease (AD) drug development. In the present study, four potent small molecule inhibitors (A01, A02, A03 and A04) of AChE with new chemical scaffold were identified. Inhibitor A03 displayed the most potent inhibition activity on AChE at enzymatic level with IC50 value of 180 nM, and high selectivity towards AChE over butyrylcholinesterase (BChE) by more than 100-fold. The binding modes of compounds A01–A04 were carefully analyzed by molecular docking and molecular dynamics (MD) simulation to provide informative clues for further structure modification. Finally, the anti-amyloid beta (Aβ) aggregation and neuroprotective activity were also well investigated. Our findings highlighted the therapeutic promise of AChE inhibitors A01–A04 for AD treatment.
Introduction
Alzheimer’s disease (AD) is the most common progressive and multifaceted neurodegenerative disease, with clinical manifestations of memory loss and cognitive impairment.1) World Alzheimer’s Report showed that in 2015 there were about 50 million Alzheimer’s patients all over the world and the number of AD cases was estimated to be 0.15 billion by 2050 as global population increasing.2)
Although the pathogenesis and etiology of AD are still unknown, the hypothesis of cholinergic dysfunction and amyloid beta (Aβ) protein aggregation were made reference to be involved in the development of AD.3) The cholinergic dysfunction hypothesis put forward that the cholinergic neurotransmission in the brain could be improved by increasing acetylcholine (ACh).4) In the brain, acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) function as two main ACh hydrolases, of which AChE is nearly 1013 fold more active than BChE by taking responsibility for 80% of the ACh activity.5) Consequently, discovery of small molecules to inhibit AChE activity has been proven as an effective approach for AD treatment. Four AChE inhibitors (AChEIs) have been used in clinic to improve memorial and cognitive function of AD patients, but tacrine was discontinued in 2013 due to safety concerns, and others (e.g., donepezil, galantamine, and rivastigmine) were unable to stop or reverse the progression of AD. In recent years, anti-AD therapies by targeting Aβ have received more attention.6) Aβ peptide is the protein composed of 39–42 amino acids, and its aggregation in the brain is believed to be an important AD pathological hallmark.7,8) It was hypothesized that Aβ aggregation could trigger critical intracellular signaling pathway resulting in nerve cells stress and apoptosis. The soluble oligomers consisting of Aβ1–42 peptide are the more toxic species.9,10) Therefore, an alternative attractive anti-AD strategy is to prevent the formation of Aβ1–42 aggregation and its toxicity towards neuronal cells.11,12) Until now, a number of dual-function agents targeting Aβ aggregation and preventing against Aβ-induced neuronal cell injury/death are in the pharma pipeline.
Multi-target drugs was believed to hold promise for the treatment of complex diseases, thus there appears a growing trend in the development of a multi-functional anti-AD drugs due to the multi-factors involved in the pathogenesis of AD. For example, a plenty of multi-functional small molecules, simultaneously functioning as AChEIs, Aβ aggregation inhibitors, and neuroprotective agents, were recently reported by our or other groups.13–20) To identify more new multifunctional AChEI candidates, we screened our in-house compound library database containing synthetic compounds, natural products and their derivatives by molecular docking-based virtual screening approach and enzyme inhibition activity screening. As a result, four new selective AChE inhibitors (numbered A01–A04) were discovered, and then their binding mode with AChE enzyme was studied by computing method. Furthermore, their inhibitory activity against Aβ aggregation and neuroprotective activity were tested. Herein, we describe the virtual screening and biological evaluation of these potential anti-AD compounds A01–A04.
Results and Discussion
Fifty Candidates Were Obtained via Molecular Docking-based Virtual ScreeningVirtual screening methods have been successfully used to identify four AChEIs with the IC50 values ranging from 1.62 to 5.73 µM by screening the SPECS database (https://www.specs.net/).18) In this work, we first re-docked the dihydrotanshinone I (1) to its crystal structure to assess the reliability of the adapted molecular docking method. As presented in Fig. S1, the re-docked pose could well reproduce the conformation in crystal structure with the rmsd value of 0.12 Å. Then the virtual screening model was used to screen our in-house database for AChEIs, and finally 50 candidates were selected for experimental validation (see Fig. S2 for virtual screening workflow).
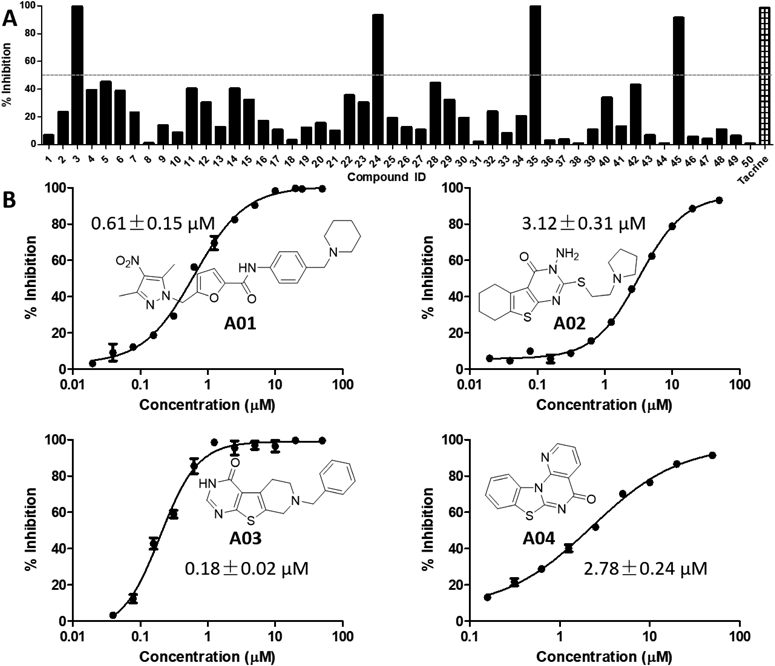
Four Potent AChEIs with New Chemical Scaffold Were Identified through in Vitro Enzymatic Inhibition Assay against AChE and BChEFirstly, the 50 potential candidates evaluated for their cholinesterase inhibitory activity at the concentration of 20 µM with tacrine as positive control. Based on the preliminary results from Fig. 1A, four compounds 3, 24, 35, 45 (correspondingly numbered as A01–A04 with structures shown in Fig. 1B) were exhibited to have >50% inhibition ratio against huAChE (human AChE) at 20 µM. Then, the IC50 values for A01–A04 were tested, and the IC50 curve for these compounds were shown in Fig. 1B. The result showed that these compounds possessed inhibitory activity against AChE with IC50 values ranging from 0.18 ± 0.02 to 3.12 ± 0.31 µM, of which A01 showed comparable activity with that of tacrine (IC50: 0.81 ± 0.11 µM) and A03 showed much better activity than tacrine. Furthermore, the selectivity of A01–A04 towards AChE was preliminary studied by testing their effects on BChE at an initial concentration of 50 µM. Interestingly, as shown in Table S1, all of them did not show obvious activity in BChE bioassay with inhibition ratio less than 50% (IC50 values for tacrine: 0.02 ± 0.01 µM), which were considered to be inactive towards BChE. Thus, these above results indicated that compounds A01–A04 were new selective AChE inhibitors. The four hits were then docked to BChE protein (Fig. S3) to probe the selectivity, as shown in Table S3, all of the four hits displayed lower docking scores with BChE compared with AChE, which in accordance with their selectivity against AChE over BChE.
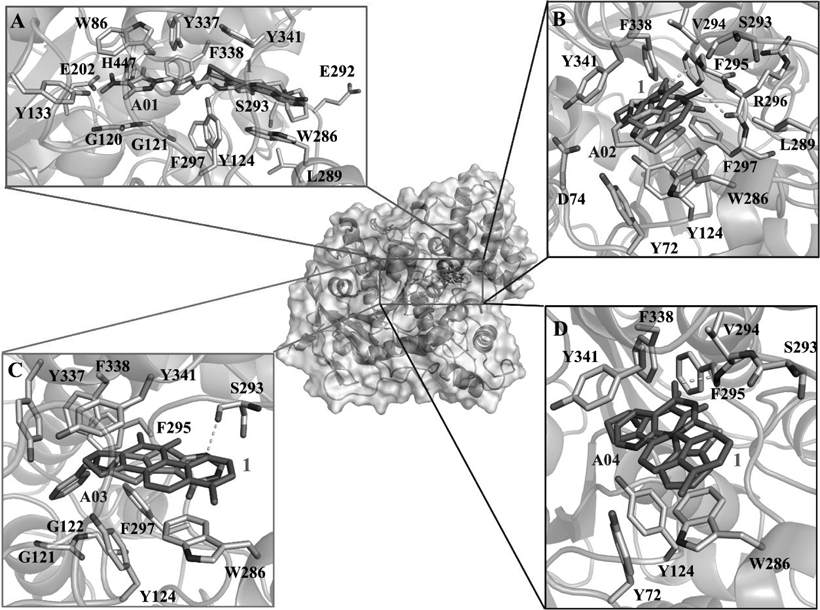
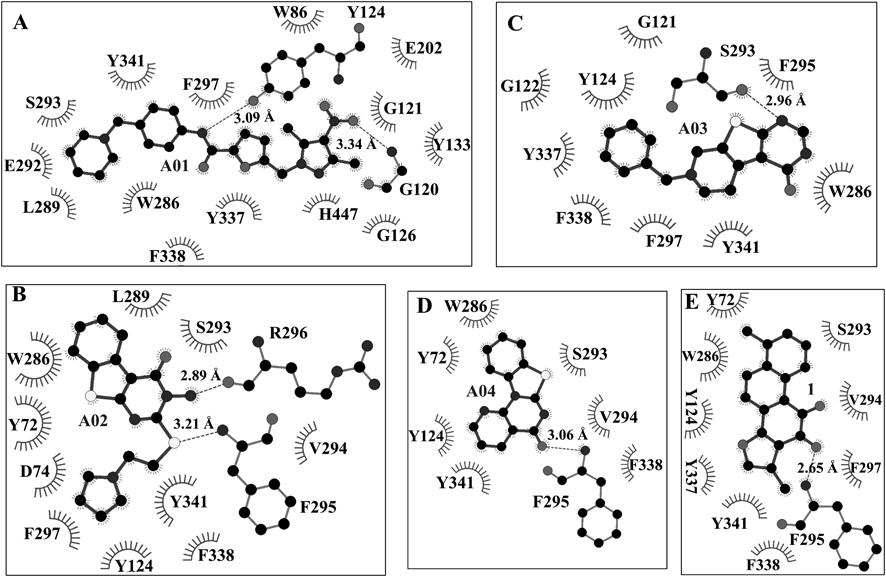
The Binding Modes of the Four Identified AChEIs Were AnalyzedTo investigate the interactions between AChE and the four AChEIs (A01–04), the probable binding modes of A01–04 with AChE were carefully analyzed based on the molecular docking results. As shown in Fig. 2, all of the four hits could well fit the dihydrotanshinone I site and displayed similar binding modes. The detailed interactions (hydrophobic and H-bond interactions) were shown in Fig. 3. From these interactions, we can see that dihydrotanshinone I binding site was a hydrophobic site, and residues Y124, W286, S293, F338, and Y341 contributed the mainly binding affinity with all of the four hits. Besides, each hit established H-bond interactions with AChE. This results provided clues for mutation assay to decrease the enzymatic activity of AChE.
Binding Modes Validation and Binding Free Energy Calculation of the Four AChEIsAfter obtaining the binding modes of the four AChEIs (A01–04) with AChE, we further performed molecular dynamics (MD) simulations to validate their binding modes. All of the four complex model (AChE with the hit) were subjected to 100 ns MD simulation. Then the root-mean-square deviations (RMSDs) values of the backbone atoms (Cα, N, C) in AChE protein and the heavy atoms in the hit of each complex model were calculated and shown in Fig. S3–S6. The small fluctuations and the hydrogen bonds occupancy analysis result (Table S2) indicated that all the four AChEIs presented well binding with AChE, and their binding modes were reliable. Subsequently, the binding free energy between the four hits and AChE was calculated via Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) method. As shown in Table 1, the order of the binding free energy of the four hits was consistent with that of the activity, which further confirmed the reliability of the predicted binding modes.
Table 1. The Binding Free Energy Calculated by MM-PBSA Method
| Compound | ΔG (kcal/mol) |
|---|
| A01 | −33.1881 ± 5.0439 |
| A02 | −18.6813 ± 3.7121 |
| A03 | −39.9766 ± 4.1223 |
| A04 | −23.2787 ± 5.5233 |
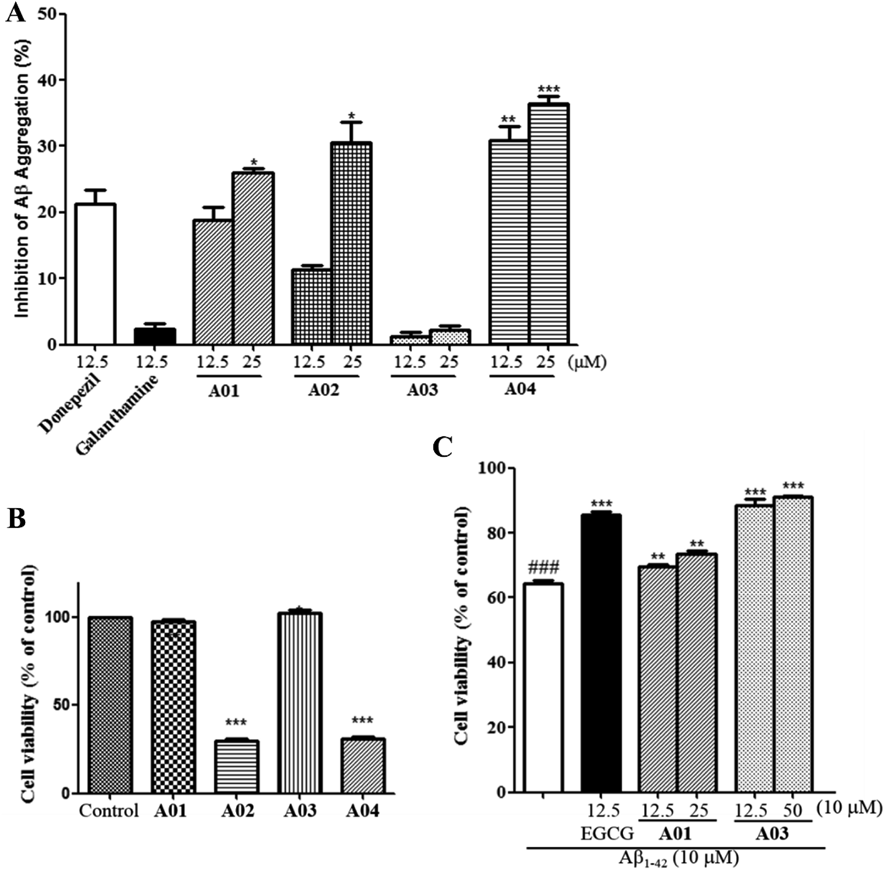
It was known that the Aβ aggregation is a key pathological hallmark of AD.7,8) AChE was considered as a nucleation factor for Aβ aggregation, and it could accelerate the formation of Aβ aggragation.21) Since lots of AChE inhibitors are able to inhibit the aggregation of Aβ, the ability of compounds A01–A04 to inhibit Aβ aggregation was evaluated using a thioflavin T (ThT) fluorometric assay,22,23) with Donepezil as positive control, and galantamine as negative control. As shown in Fig. 4A, none significant inhibitory activity was observed for the negative control galantamine at 12.5 µM. At the same screening concentration, the inhibition ratios of compounds A01, A02 and A04 against Aβ1–42 aggregation are 28.82 ± 2.03%, 11.28 ± 0.69%, 30.82 ± 2.26%, respectively, and the inhibitory ratios of these compounds rose with the screening concentration (50 µM) increasing. Among them, compound A04 showed significant anti-Aβ aggregation activity compared to donepezil at 12.5 µM (p < 0.01) and 50 µM (p < 0.001). It was worth noting that although A03 showed the highest AChE inhibitory activity, it did not exhibit activity against Aβ1–42 aggregation.
Neuroprotective Activity against Aβ1–42-Induced Neurotoxicity in SH-SY5Y CellsAs mentioned above, the aggregation of extracellular Aβ aggregation plays an critical role in the pathogenesis of AD by inducing cell stress and apoptosis of neuron cells.24) Thus, to investigate whether hits A01–A04 have neuroprotective activity, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method was perform to test the protective activity of these compounds against Aβ1–42-induced toxicity in SH-SY5Y cell models. Epigallocatechin gallate (EGCG) was used as a positive control. First, the effect of these compounds on the viability of SH-SY5Y cells was tested. The results shown in Fig. 4B indicated that A01 and A03 did not have effect on the viability of SH-SY5Y cells and, unfortunately, A02 and A04 were toxic toward SH-SY5Y at 25 µM.
Next, only compounds A01 and A03 were taken for further neuroprotective activity testing. The result of neuroprotective activity of A01 and A03 against Aβ1–42-induced toxicity are shown in Fig. 4C. In control group (white bar), the cell viability of SH-SY5Y significantly decreased to 64.30 ± 0.90% after treating by 10 µM Aβ1–42 for 24 h. In treated groups, both compounds A01 and A03 exhibited significant neuroprotective activity against Aβ induced toxicity, by increasing SH-SY5Y cell viability up to 69.74 ± 0.56% and 88.57 ± 1.93% at 12.5 µM, and 73.64 ± 0.78% and 91.02 ± 0.29% at 25 µM, respectively. Interestingly, compound A03 was inactive against Aβ1–42 aggregation but it showed comparable activity with EGCG (85.39 ± 1.00% at 12.5 µM), indicating that this compound could protect nerve cells by activating cell-protection mechanism when the cells suffered from Aβ1–42-induced injury.
Conclusion
In the present study, four (A01–04) novel scaffold AChE inhibitors were identified by molecular docking-based virtual screening. The binding modes of A01–04 with AChE were analyzed and further validated by MD simulations. The identified four potent small molecule inhibitors A01–A04 provide informative clues for further structure modification. What’s more, the inhibition on Aβ aggregation, neurotoxicity, and neuroprotective activity against Aβ-induced nerve cell injury of these compounds were well investigated. As a result, two compounds A01 and A03 were found to have ability of protecting SH-SY5Y cells against Aβ1–42-induced injury. In general, this work provided four new promising skeletons of multifunctional selective AChEIs, which deserves further chemical optimization for the development of anti-AD drug.
Experimental
Virtual ScreeningVirtual screening method used in this work was molecular docking with the Glide 7.525,26) software. The crystal structure of AChE protein complexed with an inhibitor dihydrotanshinone I (1) (PDB code: 4M0E27)) was selected to construct the molecular docking model. In brief, Protein Preparation Wizard Workfow was used to prepare AChE protein coordinates (PDB code: 4M0E) with default settings. Receptor Grid Generation panel was then carried out to generate the docking grid by defining residues located within 15 Å centered on the dihydrotanshinone I. Finally, all the prepared compounds that had been dealt with LigPrep (version 2.3, Schrödinger, LLC, New York, NY, U.S.A.) of the in-house database were docked to the aforementioned docking gird with Extra precision (XP) mode. According to the XP G-score evaluation, the top 200 candidates were selected for the following cluster analysis. Cluster analysis was performed with Pipeline Pilot 7.5 software. A similar molecular docking protocol was used as above to obtain the binding modes of A01–04 with BChE. The coordinates in PDB code 4BDS were used to perform the docking simulation of BChE.
AChE/BChE Inhibitory BioassayThe ChE inhibitory bioassay was performed by using modified Ellman’s method.28) Enzyme (AChE from human erythrocyte or BChE from equine serum) was dissolved in phosphate buffered saline (PBS) and pre-prepared at 2.0 U/mL. Ten microliters of tested compound, 20 µL of 0.01 M 5,5′-dithiobis(2-nitrobenzoic acid), 10 µL of enzyme solution, and 40 µL of PBS were co-incubated for 5 min. Then, 20 µL of 0.01 M acetylthiocholine (for AChE bioassay) or butyrylthiocholine (for BChE bioassay) was added into the well. The activity was determined by measuring the increase in absorbance at 410 nm at 37°C in 2 min intervals using Tecan Spark multimode microplate reader (Mannedorf, Switzerland). The percentage of inhibition (I) was calculated according to the formula: I = (Ac − Ai)/Ac×100%, with Ai and Ac representing the change in the absorbance in the presence of an inhibitor and without an inhibitor, respectively.29)
MD Simulation and MM-PBSA Based Binding Free Energy CalculationAMBER 14.0 software30) coupled with the Amber14SB force field was used to execute the MD simulation. The online H++ program31) was employed to predict the protonation states of all ionizable residues of AChE protein. The charges and force field parameters of A01–04 were derived from antechamber32,33) and the in general Amber force field (GAFF).32) Ligand charges of A01–04 were +1, +1, +1, and +2, respectively. The simulation model (AChE protein complexed with hit compound) was surrounded with the transferable intermolecular potential 3P water molecules (TIP3P) and neutralized with counter-ions (Na+), A01-AChE, A02-AChE, and A03-AChE simulation models were all neutralized with one Na+ ion, and A04-AChE simulation model was neutralized with two Na+ ions. MD simulations were conducted with time step of 2 fs at constant temperature and pressure with periodic boundary conditions.
When obtaining 100 ns MD trajectory, 2000 snapshots from each simulation model trajectory were extracted for MM-PBSA calculation. MM-PBSA based binding free energy calculation were carried out with MMPBSA.py.MPI module inserted in AMBER 14.0.
Anti-Aβ1–42 Aggregation BioassayThe compounds’ anti-Aβ1–42 aggregation potency was determined by using ThT-based fluorometric assay method.34) The detailed protocol was included in Supplementary Materials.
Cytotoxicity and Neuroprotective Activity Evaluation in SH-SY5Y Cell ModelThe cytotoxicity and neuroprotective activity of compound were tested in SH-SY5Y cells by using MTT method,35) which can be found in Supplementary Materials.
Statistical AnalysisAll of the experimental values were expressed as mean ± standard deviation (S.D.) of three independent experiments. Statistical analyzes were performed with GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, U.S.A.).
Acknowledgments
This research work was financially supported by the Grant from the Chia Tai Tianqing Pharmaceutical Group Co., Ltd. (YWJKJJHKYJJ-Q1115).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
References
- 1) Wiemann J., Loesche A., Csuk R., Bioorg. Chem., 74, 145–157 (2017).
- 2) Alzheimer’s Disease International, “World Alzheimer Report 2018.”: ‹https://www.alz.co.uk/research/world-report-2018›, cited on 14/7/14.
- 3) Scarpini E., Schelterns P., Feldman H., Lancet Neurol., 2, 539–547 (2003).
- 4) Bartus R. T., Dean R. L. 3rd, Beer B., Lippa A. S., Science, 217, 408–414 (1982).
- 5) Feng B., Li X., Xia J., Wu S., J. Enzyme Inhib. Med. Chem., 32, 968–977 (2017).
- 6) Kodamullil A. T., Zekri F., Sood M., Hengerer B., Canard L., McHale D., Hofmann-Apitius M., Nat. Rev. Drug Discov., 16, 819 (2017).
- 7) Yuan C., Gao Z., Chem. Res. Toxicol., 26, 262–269 (2013).
- 8) Razzokov J., Yusupov M., Bogaerts A., Sci. Rep., 9, 5476 (2019).
- 9) Selkoe D. J., Behav. Brain Res., 192, 106–113 (2008).
- 10) Caruso G., Distefano D. A., Parlascino P., Fresta C. G., Lazzarino G., Lunte S. M., Nicoletti V. G., Mol. Cell. Biochem., 425, 85–93 (2017).
- 11) Thorsett E. D., Latimer L. H., Curr. Opin. Chem. Biol., 4, 377–382 (2000).
- 12) Ahmad A., Ali T., Park H. Y., Badshah H., Rehman S. U., Kim M. O., Mol. Neurobiol., 54, 2269–2285 (2017).
- 13) Bolognesi M. L., Andrisano V., Bartolini M., Banzi R., Melchiorre C., J. Med. Chem., 48, 24–27 (2005).
- 14) Piazzi L., Rampa A., Bisi A., Gobbi S., Belluti F., Cavalli A., Bartolini M., Andrisano V., Valenti P., Recanatini M., J. Med. Chem., 46, 2279–2282 (2003).
- 15) Romero A., Marco-Contelles J., Curr. Top. Med. Chem., 17, 3328–3335 (2017).
- 16) Sharma P., Tripathi A., Tripathi P. N., Prajapati S. K., Seth A., Tripathi M. K., Srivastava P., Tiwari V., Krishnamurthy S., Shrivastava S. K., Eur. J. Med. Chem., 167, 510–524 (2019).
- 17) Goyal D., Kaur A., Goyal B., ChemMedChem, 13, 1275–1299 (2018).
- 18) Jiang C. S., Ge Y. X., Cheng Z. Q., Song J. L., Wang Y. Y., Zhu K., Zhang H., J. Comput. Aided Mol. Des., 33, 521–530 (2019).
- 19) Cheng Z. Q., Song J. L., Zhu K., Zhang J., Jiang C. S., Zhang H., Mar. Drugs, 16, 293 (2018).
- 20) Jiang C. S., Ru T., Yao L. G., Miao Z. H., Guo Y. W., Fitoterapia, 136, 104176 (2019).
- 21) Carvajal F. J., Inestrosa N. C., Front. Mol. Neurosci., 4, 19 (2011).
- 22) Bartolini M., Bertucci C., Bolognesi M. L., Cavalli A., Melchiorre C., Andrisano V., ChemBioChem, 8, 2152–2161 (2007).
- 23) Ha H. J., Kang D. W., Kim H. M., Kang J. M., Ann J., Hyun H. J., Lee J. H., Kim S. H., Kim H., Choi K., Hong H. S., Kim Y., Jo D. G., Lee J., Lee J., J. Med. Chem., 61, 396–402 (2018).
- 24) Maher P., Davis J. B., J. Neurosci., 16, 6394–6401 (1996).
- 25) Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem., 47, 1750–1759 (2004).
- 26) Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem., 47, 1739–1749 (2004).
- 27) Cheung J., Gary E. N., Shiomi K., Rosenberry T. L. ACS Med. Chem. Lett., 4, 1091–1096 (2013).
- 28) Talesa V. N., Mech. Ageing Dev., 122, 1961–1969 (2001).
- 29) Beesu M., Caruso G., Salyer A. C., Khetani K. K., Sil D., Weerasinghe M., Tanji H., Ohto U., Shimizu T., David S. A., J. Med. Chem., 58, 7833–7849 (2015).
- 30) Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., Woods R. J., J. Comput. Chem., 26, 1668–1688 (2005).
- 31) Gordon J. C., Myers J. B., Folta T., Shoja V., Heath L. S., Onufriev A., Nucleic Acids Res., 33 (Web Server), W368–W371 (2005).
- 32) Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., J. Comput. Chem., 25, 1157–1174 (2004).
- 33) Wang J., Wang W., Kollman P. A., Case D. A., J. Mol. Graph. Model., 25, 247–260 (2006).
- 34) Fang L., Fang X., Gou S., Lupp A., Lenhardt I., Sun Y., Huang Z., Chen Y., Zhang Y., Fleck C., Eur. J. Med. Chem., 76, 376–386 (2014).
- 35) Mei W. W., Ji S. S., Xiao W., Wang X. D., Jiang C. S., Ma W. Q., Zhang H. Y., Gong J. X., Guo Y. W., Monatshefte für Chemie-Chemical Monthly, 148, 9 (2017).