Introduction
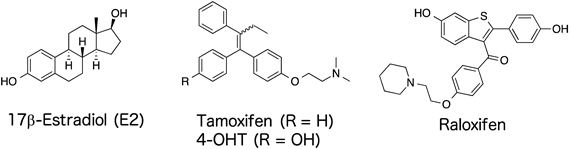
Estrogen receptor (ER), which is a superfamily of nuclear receptors, plays pivotal roles in physiological actions, such as cell growth and bone marrow homeostasis.1,2) 17β-Estradiol, an endogenous ligand for ERα, binds to the ligand binding domain of ERα and promotes its transactivation, resulting in target gene transcription. It has been reported that ERα is overexpressed in 70% of breast cancer cells and that it plays an important role in promoting tumor progression.3) Therefore, ERα may be a promising target for breast cancer therapy, and the several ERα antagonists have been developed4–6) (Fig. 1). Among these, tamoxifen and raloxifen are well known as selective ER modulators and broadly used for breast cancer therapy.6)
Although tamoxifen shows promising antitumor effects against breast cancer cells, it also acts as an agonist against in the uterus ERα,7) and such poor organ selectivity become a problem in its clinical use. To overcome poor organ selectivity, innovative strategies have been urgently required for development of novel ligands. Recently, photopharmacology has garnered attention as a promising strategy to regulate the bioactivity of compounds under UV irradiation. To data, several photopharmaceutical agents containing diazo, spiropyran, and diarylethene moieties have been reported8–10) (Fig. 2). Especially, compounds that contain a diazo moiety undergo trans-to-cis isomerization under irradiation with light of an appropriate wavelength.8) Owing to its photochemical properties, the azobenzene moiety has been broadly utilized to change the conformation of compounds and control their bioactivities.11)
With the aim of development of novel ER ligands, we hypothesized that photopharmacological ER ligands are a promising strategy to overcome the organ selectivity of ER ligands in the treatment of breast cancer. Recently, photoswitchable Farnesoid X receptor (FXR) ligands based on GW4064, a representative FXR agonist, have been reported.12) The compounds have a diazo moiety instead of the unsaturated double bond of GW4064, and their FXR activity is regulated by UV irradiation in cells. These findings suggested that photoswitchable reagents are applicable for intracellular proteins. Previously, Hashimoto and colleagues reported that diphenylmethane (DPM) skeleton can be a bioisoster of steroid skeleton, and appropriate modifications of DPM skeleton can be applied to develop several nuclear receptor ligands, such as vitamin D receptor and ERs.13,14) Among these ligands, PBP showed potent antagonistic activity against ERα, with IC50 value of 4.9 nM.13) Furthermore, its diphenylsilane derivative also exhibits transactivation activity against ERα, with IC50 value of 29.7 nM,15) indicating that substitution of heptane chain with other atoms is applicable in the development of ER ligands. Hence, we designed and synthesized a set of photoswitchable ER ligands containing a diazo moiety (Fig. 3). Moreover, we evaluated their UV spectra to confirm their conformational change after UV irradiation, and we investigated the binding of the synthesized compounds to ERα.
Results
ChemistryInitially, we prepared a set of azobenzene derivatives 1–10, via coupling and modification of aryl diazonium species with phenolic compounds (Chart 1). Azobenzene 1, 2, 5, 8 and 9 were synthesized according to a previously reported method.16–20) Next, the phenolic hydroxy group on 1 and 8 was alkylated to afford 2–6 and 9, and both hydroxy groups in 1 were acetylated (7). To synthesize tetrafluoroazobenzene 8 and 9, 2,6-difluoroaniline and (4-amino-3,5-difluorophenyl)methanol were converted to diazonium salt via treatment with NaNO2 in conc. HCl. Next, the resultant diazonium salt was subjected to a coupling reaction with 3,5-difluorophenol in the presence of aqueous NaOH, thus yielding tetrafluoroazobenzene with or without a hydroxymethyl group; methylation of a phenolic hydroxy group of compound 8 provided 9 as a mixture of E- and Z-isomers, but isomerization of Z- to E-isomer occurred during column chromatography.

Chart 1. Synthesis of Compounds 1–10
Next, we evaluated the conformational changes in compounds 1–10 by measuring the UV spectra and the E/Z ratio of the compounds were determined using HPLC analysis. The UV spectra of compounds 1–10 are shown in Fig. 4. Compound 1 did not exhibit any conformational change after UV irradiation at 254 or 365 nm, whereas compounds 2, 6, and 7 showed dramatic conformational changes from E- to Z-form after UV irradiation at 365 nm. These results showed that alkylation of the phenol group may exert an advantage to their conformational change. However, further investigation revealed that the Z-isomer of compounds 2–7 was rapidly reversed to the E-form at room temperature (25°C) even in the dark; therefore, the effects of these conformational changes on the binding affinity of the compounds to ERα could not be investigated. It has been reported that o-fluorination of diphenyldiazo compounds can stabilize and prolong the E/Z-conformation via their steric effect.21) On the basis of this report, we employed tetra-fluorinated diphenyldiazo compounds at the o-position, and confirmed their conformational change after UV irradiation. Compound 8 did not show any conformational change after UV irradiation, whereas compounds 9 and 10, which had four fluorine atoms at the o-position showed conformational changes after UV irradiation (Table 1; E/Z ratio: 9, 13/87; 10, 11/89), and their half-life (t1/2) in the E-form was prolonged because of their steric effects (Table 1; E/Z ratio after 16 h incubation at 37°C: 9, 28/72; 10, 32/68). These data revealed that the phenolic hydroxy group at the p-position took disadvantages on conformational control by UV irradiation, and the compounds 9, and 10 could be a candidate as photoswitchable ERα ligands.
Table 1.
E/
Z Ratio of Compounds
9 and
10 | E/Z-Area ratio |
|---|
| E-Isomer | Z-Isomer |
|---|
| (E)-9 Nature | 89% | 11% |
| (Z)-9 365 nm 3 min | 13% | 87% |
| (Z)-9 37°C 16 h | 28% | 72% |
| (E)-10 Nature | 94% | 6% |
| (Z)-10 365 nm 3 min | 11% | 89% |
| (Z)-10 37°C 16 h | 32% | 68% |
Finally, to investigate the effects of conformational changes on the binding affinity of the compounds to ERα, the binding of compounds 9 and 10 to ERα was examined. The binding affinity of 9 and 10 was evaluated using fluorescent polarization (FP) assay kit following the manufacturer’s instruction (Thermo Fisher Scientific, U.S.A.). As shown in Fig. 5, Z-isomer of compound 9 showed moderate binding affinity to ERα, whereas its E-isomer had no binding affinity even at 10 µM. Moreover, the Z- and E-isomers of compounds 10 bound to ERα, but the binding affinity of compounds 10 was lower than that of 9. These results showed that the Z-isomer of compounds 9 and 10 exhibited the higher binding affinity to ERα than did the E-isomer of both compounds, and that introduction of a hydroxymethyl group at the p-position decreased the binding affinity of the compounds to ERα.
In conclusion, we designed and synthesized a set of photoswitchable ER ligands based on diphenyldiazo skeleton. Introduction of fluorine at the o-position prolonged their half-life (t1/2) as a Z-form by its steric effects. Moreover, the binding affinity of compounds to ERα was investigated using FP assay, and the results revealed that the Z-isomer of compound 9 showed higher binding affinity to ERα than did the E-isomer. Although the binding affinity of compound 9 was moderate compared to that of known ER ligands, such as tamoxifen, the ER binding affinity was controlled by UV irradiation, suggesting that compound 9 can be a promising candidate of a photoswitchable ERα ligand. The detailed structure–activity relationship (SAR) analysis and ERα-transactivation activity of compound 9 in the cells are in progress.
Experimental
GeneralAll chemicals were purchased from Sigma-Aldrich (U.S.A.), FUJIFILM Wako Pure Chemical Corporation (Japan), and Tokyo Chemical Industry (Japan) and were used without further purification. TLC analysis was conducted using Merck silica gel 60 F254 pre-coated plates and visualized using a 254 nm/365 nm UV lamp, and iodine stain. Column chromatography was performed using silica gel (spherical, neutral) purchased from Kanto Chemical. 1H- and 13C-NMR spectra were recorded on a JEOL ECZ600 spectrometer, and measurements were carried out in CDCl3 with 0.03% tetramethylsilane or dimethyl sulfoxide (DMSO)-d6. High-resolution mass spectra (HR-MS) were measured using a Shimadzu IT-TOF MS equipped with an electrospray ionization source.
(4-((4-Ethoxyphenyl)diazenyl)phenyl)methanol (3)To a solution of 4-(4-hydroxyphenyl)azobenzyl alcohol (1, 57.1 mg, 0.250 mmol) in N,N-dimethylformamide (DMF) (0.5 mL) were added K2CO3 (69.1 mg, 0.500 mmol) and iodoethane (22.2 µL, 0.275 mmol) at room temperature, and the mixture was warmed to 70°C. After being stirred for 4 h, the reaction mixture was diluted with EtOAc/n-hexane (1 : 3), and washed successively with 1 M HCl and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (50% EtOAc in n-hexane) to give title compound 3 (44.1 mg, 69%) as an orange solid. mp 126–128°C; 1H-NMR (600 MHz, CDCl3) δ: 1.45 (2H, t, J = 7.8 Hz), 2.03 (1H, br s, 1H), 4.10 (2H, q, J = 7.8 Hz, 2H), 4.74 (2H, s), 6.99 (2H, d, J = 8.4 Hz), 7.46 (2H, d, J = 8.4 Hz), 7.85 (2H, d, J = 6.6 Hz), 7.90 (2H, d, J = 6.6 Hz). 13C-NMR (151 MHz, CDCl3) δ: 14.72, 63.81, 64.85, 114.66, 122.71, 124.72, 127.41, 143.08, 146.83, 152.20, 161.48. HR-MS (electrospray ionization (ESI)) m/z: [M + H]+ Calcd for C15H17N2O2, 257.1285. Found, 257.1276.
(4-((4-n-Propylphenyl)diazenyl)phenyl)methanol (4)To a solution of 4-(4-hydroxyphenyl)azobenzyl alcohol (1, 57.1 mg, 0.250 mmol) in DMF (0.5 mL) were added K2CO3 (69.1 mg, 0.500 mmol) and 1-iodopropane (26.8 µL, 0.276 mmol) at room temperature, and the mixture was warmed to 70°C. After being stirred for 4 h, the reaction mixture was diluted with EtOAc/n-hexane (1 : 3), and washed successively with 1 M HCl and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (50% EtOAc in n-hexane) to give title compound 4 (44.7 mg, 66%) as an orange solid. mp 130°C; 1H-NMR (600 MHz, CDCl3) δ: 1.05 (3H, t, J = 6.6 Hz), 1.84 (2H, sextet, J = 6.6 Hz), 2.16 (1H, br s), 3.98 (2H, t, J = 6.6 Hz), 4.72 (2H, s), 6.99 (2H, d, J = 8.4 Hz), 7.45 (2H, d, J = 8.4 Hz), 7.84 (2H, d, J = 7.2 Hz), 7.89 (2H, d, J = 7.2 Hz). 13C-NMR (151 MHz, CDCl3) δ: 10.45, 22.47, 64.79, 69.79, 114.67, 122.68, 124.69, 127.38, 143.07, 146.78, 152.16, 161.67.; HR-MS (ESI) m/z: [M + H]+ Calcd. for C16H19N2O2, 271.1441. Found, 271.1434.
(4-((4-(2-(Dimethylamino)ethoxy)phenyl)diazenyl)phenyl)methanol (6)To a solution of 4-(4-hydroxyphenyl)azobenzyl alcohol (1, 113 mg, 0.499 mmol) and K2CO3 (1.43 g, 10.3 mmol) in acetone/water (9 : 1, 5 mL) were added 2-(dimethylamino)ethyl chloride hydrochloride (262 mg, 1.82 mmol) and K2CO3 (390 mg, 2.82 mmol) at 0°C, and the mixture was stirred at the same temperature for 30 min. Then, the mixture was refluxed for 2 h. After cooling to room temperature, the solution was filtered, and the filtrate was concentrated in vacuo. To the residue was added Et2O, and the suspension was filtered to afford the title compound 6 (102 mg, 68%) as an orange solid. mp 142–144°C; 1H-NMR (600 MHz, DMSO-d6) δ: 2.19 (6H, s), 2.61 (2H, t, J = 6.0 Hz), 4.11 (2H, t, J = 6.0 Hz), 4.55 (2H, s), 5.35 (1H, s), 7.09 (2H, d, J = 8.4 Hz), 7.46 (2H, d, J = 8.4 Hz), 7.78 (2H, d, J = 7.2 Hz), 7.83 (2H, d, J = 7.2 Hz). 13C-NMR (151 MHz, DMSO-d6) δ: 45.54, 57.56, 62.45, 66.27, 115.05, 122.18, 124.44, 127.08, 145.65, 146.09, 150.89, 161.15. HR-MS (ESI) m/z: [M + H]+ Calcd for C17H22N3O2, 300.1707. Found, 300.1714.
4-((4-(Acetoxymethyl)phenyl)diazenyl)phenyl Acetate (7)To a solution of 4-(4-hydroxyphenyl)azobenzyl alcohol (1, 50.0 mg, 0.219 mmol) in pyridine (2.2 mL) were added Et3N (61.4 µL, 0.441 mmol), N,N-dimethyl-4-aminopyridine (13.5 mg, 0.111 mmol) and acetic anhydride (23.0 µL, 0.243 mmol) at 0°C, and the mixture was gradually warmed to room temperature. After being stirred for 6 h, the reaction mixture was diluted with EtOAc, and washed successively with 1 M HCl and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (5–20% EtOAc in n-hexane) to give title compound 7 (11.6 mg, 17%) as an orange solid. mp 100–102°C; 1H-NMR (600 MHz, CDCl3) δ: 2.07 (3H, s), 2.27 (3H, s), 5.11–4.96 (2H, m), 7.18 (2H, d, J = 8.4 Hz), 7.43 (2H, d, J = 8.4 Hz), 7.83 (2H, d, J = 6.6 Hz), 7.89 (2H, d, J = 6.6 Hz). 13C-NMR (151 MHz, CDCl3) δ: 20.98, 21.16, 65.69, 122.23, 124.10, 123.02, 128.76, 138.85, 150.17, 152.23,152.71, 169.10, 170.79. HR-MS (ESI) m/z: [M + H]+ Calcd for C17H17N2O4, 313.1183. Found, 313.1189.
(4-((1,6-Difluro-4-methoxyphenyl)diazenyl)-3,5-difluorophenyl)methanol (10; E/Z-Isomer)To a solution of (4-amino-3,5-difluorophenyl)methanol (32.7 mg, 0.205 mmol) and conc. HCl (100 µL) in water (750 µL) was added dropwise NaNO2 (15.6 mg, 0.226 mmol) in water (450 µL) at 0°C. Then, a cooled solution of 3,5-difluorophenol (29.3 mg, 0.225 mmol) in 1 M NaOH aq was added dropwise to the reaction mixture, and the mixture was stirred at the same temperature for 1 h. After acidified with 1 M HCl, the solution was extracted with EtOAc, and the combined organic phase was dried over MgSO4. Removal of the solvent afforded a crude product, which was purified by column chromatography on silica gel (40% EtOAc in n-hexane) to give 3,5-difluoro-4-(2,6-difluoro-4-hydroxyphenyl)azobenzyl alcohol (S-1, 15.8 mg, 26%). To a solution of S-1 (15.8 mg, 52.6 µmol) in DMF (0.5 mL) were added K2CO3 (14.5 mg, 0.105 mmol) and iodomethane (4.26 µL, 68.4 µmol) at room temperature, and the mixture was warmed to 70°C. After being stirred for 5 h, the reaction mixture was diluted with EtOAc/n-hexane (1 : 2), and washed successively with 1 M HCl and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (25% EtOAc in n-hexane) to give title compound 10 (E,Z-mixture: 8.0 mg, 48%, NOTE: A part of Z-isomer isomerized to E-isomer during flash column chromatography) as an orange solid. mp 152–154°C; 1H-NMR (600 MHz, CDCl3) δ: 3.88–3.77 (3H, m), 4.75–4.66 (2H, m), 6.61–6.37 (2H, m), 7.06–6.88 (2H, m). 13C-NMR (151 MHz, CDCl3) δ: 55.97, 56.17, 63.62, 63.81, 98.36 (d, J = 23.1 Hz), 98.91 (d, J = 23.1 Hz), 109.70 (d, J = 20.2 Hz), 110.08 (d, J = 20.2 Hz), 126.02–125.87 (m), 131.01–130.82 (m), 143.65 (t, J = 8.6 Hz), 144.89 (t, J = 8.6 Hz), 151.79 (dd, J = 252.8, 5.7 Hz), 152.86 (dd, J = 252.8, 7.2 Hz), 155.62 (dd, J = 261.5, 5.7 Hz), 157.45 (dd, J = 261.5, 7.2 Hz), 160.90 (t, J = 13.0 Hz), 162.53 (t, J = 13.0 Hz). HR-MS (ESI) m/z: [M + H]+ Calcd for C14H10F4N2O2, 315.0751. Found, 315.0755.
UV IrradiationAS ONE Handy UV Lamp SLUV-4 254/365 nm was used as UV light source and UV spectra were measured on Jasco V-730 Spectrophotometer. Ten milli molar compounds in DMSO were dissolved in EtOH (final concentration 1 mM). UV irradiation was evaluated 254 nm for 3 min, followed by 365 nm in the same solution. E/Z ratio of 1 mM compounds were measured on HPLC after preservations at room temperature and 37°C for 16 h.
Fluorescence Polarization AssayThe relative binding affinities of tested compounds for ERα were determined by fluorescence polarization-based competition binding assay using commercially available kits (P2698, Life Technologies, Inc.) according to the manufacturer’s instruction. After 1 h, fluorescence polarization signals (mP values) were measured using the plate reader (EnVision 2105, PerkinElmer, Inc.) with a 480 nm excitation/535 nm emission filter. The fraction of compounds bound to ERα was correlated to the mP value and plotted against values of competitor concentration.