Experimental
Chemistry1H-NMR spectra were recorded on a Varian VNS-400, Varian 400-MR, JEOL JNM Lambda-300, JEOL JNM Lambda-400 or Bruker AVANCE III-HD500 spectrometer. Chemical shifts were expressed in δ values (ppm) using tetramethylsilane as the internal standard (s = singlet, d = doublet, t = triplet, q = quartet, dd = double doublet, ddd = double double doublet, m = multiplet and br = broad peak). MS were recorded on a JEOL GC Mate II, Waters SQD, Waters ZQ-2000, Thermo Fisher LCQ Advantage or Thermo Fisher Exactive Plus Orbitrap. All reactions were performed using commercially available reagents and solvents without further purification. The following abbreviations are used: AcOH, acetic acid; Boc, tert-butoxycarbonyl; BuLi, butyllithium; t-BuOK, potassium tert-butoxide; DEAD, diethyl (E)-diazene-1,2-dicarboxylate; DMF, N,N-dimethylformamide; DIPEA, N,N-diisopropylethylamine; DMSO, dimethyl sulfoxide; ESI, electrospray ionization; Et3N, triethylamine; Et2O, diethylether; EtOAc, ethyl acetate; EtOH, ethanol; HOBt, 1H-benzotriazol-1-ol; IPA, isopropyl alcohol; MeCN, acetonitrile; MeOH, methanol; NaBH(OAc)3, sodium triacetoxyborohydride; PPh3, triphenylphosphine; THF, tetrahydrofuran; p-TsOH·H2O, 4-methylbenzenesulfonic acid monohydrate; WSC·HCl, N-[3-(dimethylamino)propyl]-N′-ethylcarbodiimide hydrochloride.
1-[(4-Methoxyphenyl)methyl]piperidine-4-carboxylic Acid (3)To a solution of 4-methoxybenzyl chloride (1.1 mL, 7.7 mmol) in DMF (10 mL) was added ethyl piperidine-4-carboxylate (2; 3.0 mL, 19 mmol). After stirring at room temperature for 16 h, the mixture was diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O, saturated NaHCO3 aqueous solution and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue in THF (15 mL) and EtOH (10 mL) was added 4.0 mol/L NaOH aqueous solution (13 mL, 52 mmol). After stirring at room temperature for 3 d, the mixture was acidified with concentrated HClaq. (approximately pH 5) and concentrated in vacuo. To the residue was added CHCl3/MeOH (2 : 1), the mixture was filtered and the filtrate was concentrated in vacuo. To the residue was added EtOAc, and the precipitated solid was collected by filtration to obtain the product (1.2 g, 63%). 1H-NMR (DMSO-d6) δ: 1.46–1.63 (2H, m), 1.69–1.88 (2H, m), 1.90–2.30 (3H, m), 2.64–2.88 (2H, m), 3.08–3.63 (2H, m), 3.73 (3H, s), 6.88 (2H, d, J = 8.4 Hz), 7.21 (2H, d, J = 8.4 Hz); MS m/z: 250 (M + H)+.
(5-Aminopyridin-2-yl)(4-{[4-(trifluoromethyl)phenyl]methyl}piperazin-1-yl)methanone (5)To a solution of 5-aminopyridine-2-carboxylic acid (4; 200 mg, 1.4 mmol) in DMF (3.3 mL) were added 1-{[4-(trifluoromethyl)phenyl]methyl}piperazine (360 mg, 1.5 mmol), WSC·HCl (330 mg, 1.7 mmol) and HOBt (250 mg, 1.9 mmol). After stirring at room temperature overnight, the mixture was diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O, saturated NaHCO3 aqueous solution and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH) to obtain the product (420 mg, 79%). 1H-NMR (CDCl3) δ: 2.32–2.69 (4H, m), 3.58 (2H, s), 3.72–3.84 (4H, m), 3.90 (2H, s), 7.01 (1H, dd, J = 2.7, 8.4 Hz), 7.46 (2H, d, J = 8.0 Hz), 7.54–7.60 (3H, m), 7.98 (1H, d, J = 2.7 Hz); MS m/z: 365 (M + H)+.
1-[(4-Methoxyphenyl)methyl]-N-[6-(4-{[4-(trifluoromethyl)phenyl]methyl}piperazine-1-carbonyl)pyridin-3-yl]piperidine-4-carboxamide Trihydrochloride (6)To a solution of 1-[(4-methoxyphenyl)methyl]piperidine-4-carboxylic acid (3; 110 mg, 0.45 mmol) in CH2Cl2 (4.0 mL) were added (COCl)2 (44 µL, 0.51 mmol) and DMF (1 drop) at 0°C. After stirring at room temperature for 1 h, to the mixture were added a solution of (5-aminopyridin-2-yl)(4-{[4-(trifluoromethyl)phenyl]methyl}piperazin-1-yl)methanone (5; 140 mg, 0.37 mmol) in CH2Cl2 (2.0 mL) and pyridine (76 µL, 0.95 mmol) at 0°C. After stirring at room temperature for 4 h, the mixture was diluted with H2O and extracted with CHCl3. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a mixture of the residue in MeOH (3.0 mL) was added 4.0 mol/L HCl in 1,4-dioxane (0.50 mL, 2.0 mmol). After stirring at room temperature for 15 min, the mixture was concentrated in vacuo. To the residue was added Et2O, and the precipitated solid was collected by filtration to obtain the product (110 mg, 40%). 1H-NMR (CD3OD) δ: 1.93–2.36 (4H, m), 2.73–2.86 (1H, m), 3.03–3.16 (2H, m), 3.20–3.74 (10H, m), 3.83 (3H, s), 4.28 (2H, s), 4.52 (2H, s), 7.00–7.07 (2H, m), 7.43–7.48 (2H, m), 7.77–7.86 (5H, m), 8.28 (1H, dd, J = 2.4, 8.6 Hz), 8.94 (1H, d, J = 2.4 Hz); MS m/z: 596 (M + H)+; ESI-MS m/z: 596.2836 (M + H)+ (Calcd for C32H37O3N5F3: 596.2843).
Methyl 5-Hydroxypyridine-2-carboxylate (8)To a solution of 5-hydroxypyridine-2-carboxylic acid (7; 10 g, 72 mmol) in MeOH (120 mL) was added concentrated H2SO4 (8.0 mL, 150 mmol). The mixture was stirred at 85°C overnight. After cooling to 0°C, the mixture was basified with 1.0 mol/L NaOH aqueous solution (approximately pH 9), and then acidified with 10% citric acid aqueous solution (approximately pH 5). The mixture was extracted with CHCl3/IPA (4 : 1), the organic layer was dried over MgSO4 and concentrated in vacuo to obtain the product (8.4 g, 76%). 1H-NMR (DMSO-d6) δ: 3.82 (3H, s), 7.27 (1H, dd, J = 2.8, 8.6 Hz), 7.94 (1H, dd, J = 0.5, 8.6 Hz), 8.22 (1H, dd, J = 0.5, 2.8 Hz), 10.81 (1H, s); MS m/z: 154 (M + H)+.
5-{[1-(tert-Butoxycarbonyl)piperidin-4-yl]methoxy}pyridine-2-carboxylic Acid (9)To a solution of methyl 5-hydroxypyridine-2-carboxylate (8; 2.0 g, 13 mmol), tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate (3.0 g, 14 mmol) and PPh3 (5.0 g, 19 mmol) in THF (40 mL) was added dropwise 2.2 mol/L DEAD in toluene (9.0 mL, 20 mmol) at 0°C. After stirring at room temperature overnight, the mixture was concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/EtOAc). To a solution of the residue in THF (20 mL) and MeOH (20 mL) was added 1.0 mol/L NaOH aqueous solution (40 mL, 40 mmol). The mixture was stirred at 60°C for 1 h. After cooling to room temperature, the mixture was acidified with 10% citric acid aqueous solution (approximately ~pH 5). The mixture was extracted with CHCl3/IPA (4 : 1), the organic layer was dried over MgSO4 and concentrated in vacuo to obtain the product (4.4 g, 99%). 1H-NMR (DMSO-d6) δ: 1.10–1.25 (2H, m), 1.40 (9H, s), 1.70–1.81 (2H, m), 1.91–2.05 (1H, m), 2.60–2.91 (2H, m), 3.91–4.09 (4H, m), 7.50 (1H, dd, J = 2.8, 8.6 Hz), 8.01 (1H, d, J = 8.6 Hz), 8.36 (1H, d, J = 2.8 Hz), 8.97 (1H, s); MS m/z: 337 (M + H)+.
{5-[(Piperidin-4-yl)methoxy]pyridin-2-yl}(4-{[4-(trifluoromethyl)phenyl]methyl}piperazin-1-yl)methanone (10)To a mixture of 5-{[1-(tert-butoxycarbonyl)piperidin-4-yl]methoxy}pyridine-2-carboxylic acid (9; 1.4 g, 4.2 mmol), 1-{[4-(trifluoromethyl)phenyl]methyl}piperazine (0.85 mL, 4.3 mmol) and CH2Cl2 (25 mL) were added WSC·HCl (1.1 g, 5.7 mmol) and HOBt (750 mg, 5.6 mmol). After stirring at room temperature overnight, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue in MeOH (10 mL) was added 4.0 mol/L HCl in 1,4-dioxane (10 mL, 40 mmol). After stirring at room temperature for 2.5 h, the mixture was basified with saturated NaHCO3 aqueous solution and extracted with CHCl3/IPA (4 : 1). The organic layer was dried over MgSO4 and concentrated in vacuo to obtain the product (1.7 g, 88%). 1H-NMR (DMSO-d6) δ: 1.10–1.24 (2H, m), 1.64–1.74 (2H, m), 1.77–1.93 (1H, m), 2.24–2.57 (6H, m), 2.91–3.02 (2H, m), 3.32–3.73 (6H, m), 3.92 (2H, d, J = 6.4 Hz), 7.47 (1H, dd, J = 2.9, 8.8 Hz), 7.52–7.58 (3H, m), 7.69 (2H, d, J = 8.2 Hz), 8.24 (1H, dd, J = 0.7, 2.9 Hz); MS m/z: 463 (M + H)+.
{4-[(4-Chlorophenyl)methyl]piperazin-1-yl}{5-[(piperidin-4-yl)methoxy]pyridin-2-yl}methanone (11)To a mixture of 5-{[1-(tert-butoxycarbonyl)piperidin-4-yl]methoxy}pyridine-2-carboxylic acid (9; 3.0 g, 8.8 mmol), benzyl piperazine-1-carboxylate (1.8 mL, 9.3 mmol) and CH2Cl2 (50 mL) were added WSC·HCl (2.3 g, 12 mmol) and HOBt (1.6 g, 12 mmol). After stirring at room temperature overnight, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue in EtOH (40 mL) was added 10% Pd/C (wetted with approx. 50% water, 1.0 g). After stirring under a hydrogen atmosphere (1.0 kgf/cm2) at room temperature for 3 d, to the mixture was added 10% Pd/C (wetted with ca. 50% water, 1.5 g). After stirring under a hydrogen atmosphere (1.0 kgf/cm2) at room temperature for 2 d, the mixture was filtered through a Celite pad and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH) to obtain the intermediate (1.2 g). To a solution of the obtained intermediate (600 mg, 1.5 mmol) in CH2Cl2 (12 mL) were added 4-chlorobenzaldehyde (240 mg, 1.7 mmol), acetic acid (10 µL, 0.17 mmol) and NaBH(OAc)3 (800 mg, 3.8 mmol). After stirring at room temperature overnight, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue in MeOH (6.0 mL) was added 4.0 mol/L HCl in 1,4-dioxane (6.0 mL, 24 mmol). After stirring at room temperature for 1 h, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3/IPA (4 : 1). The organic layer was dried over MgSO4 and concentrated in vacuo to obtain the product (640 mg, 33%). 1H-NMR (DMSO-d6) δ: 1.10–1.25 (2H, m), 1.64–1.72 (2H, m), 1.76–1.91 (1H, m), 2.26–2.55 (6H, m), 2.91–3.00 (2H, m), 3.41–3.69 (6H, m), 3.91 (2H, d, J = 6.4 Hz), 7.29–7.42 (4H, m), 7.46 (1H, dd, J = 2.9, 8.8 Hz), 7.55 (1H, dd, J = 0.7, 8.8 Hz), 8.24 (1H, dd, J = 0.7, 2.9 Hz); MS m/z: 429 (M + H)+.
4-[(4-{5-[(Piperidin-4-yl)methoxy]pyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (12)Compound 12 was prepared from 9 and 4-formylbenzonitrile in 33% yield using a similar approach to that described for 11. 1H-NMR (DMSO-d6) δ: 1.10–1.29 (2H, m), 1.64–1.73 (2H, m), 1.75–1.92 (1H, m), 2.30–2.56 (6H, m), 2.90–3.01 (2H, m), 3.48–3.68 (6H, m), 3.92 (2H, d, J = 6.4 Hz), 7.44–7.59 (4H, m), 7.76–7.83 (2H, m), 8.24 (1H, dd, J = 0.4, 2.4 Hz); MS m/z: 420 (M + H)+.
[5-({1-[(4-Methoxyphenyl)methyl]piperidin-4-yl}methoxy)pyridin-2-yl](4-{[4-(trifluoromethyl)phenyl]methyl}piperazin-1-yl)methanone Trihydrochloride (13)To a solution of {5-[(piperidin-4-yl)methoxy]pyridin-2-yl}(4-{[4-(trifluoromethyl)phenyl]methyl}piperazin-1-yl)methanone (10; 300 mg, 0.65 mmol) in CH2Cl2 (6.0 mL) were added 4-methoxybenzaldehyde (90 µL, 0.74 mmol), acetic acid (5.0 µL, 87 µmol) and NaBH(OAc)3 (300 mg, 1.4 mmol). After stirring at room temperature overnight, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue in MeOH was added excess 4.0 mol/L HCl in 1,4-dioxane. The mixture was concentrated in vacuo. To the residue was added Et2O, and the precipitated solid was collected by filtration to obtain the product (230 mg, 51%). 1H-NMR (DMSO-d6) δ: 1.54–2.15 (5H, m), 2.75–3.73 (10H, m), 3.78 (3H, s), 3.91–4.06 (2H, m), 4.11–4.37 (3H, m), 4.38–4.68 (3H, m), 7.01 (2H, d, J = 8.8 Hz), 7.43–7.61 (3H, m), 7.68 (1H, d, J = 8.8 Hz), 7.78–7.97 (4H, m), 8.27 (1H, d, J = 2.9 Hz), 10.52 (1H, br s), 11.89 (1H, br s); MS m/z: 583 (M + H)+; ESI-MS m/z: 583.2888 (M + H)+ (Calcd for C32H38O3N4F3: 583.2891).
[5-({1-[(6-Methoxypyridin-3-yl)methyl]piperidin-4-yl}methoxy)pyridin-2-yl](4-{[4-(trifluoromethyl)phenyl]methyl}piperazin-1-yl)methanone Ditosylate (14)To a solution of {5-[(piperidin-4-yl)methoxy]pyridin-2-yl}(4-{[4-(trifluoromethyl)phenyl]methyl}piperazin-1-yl)methanone (10; 300 mg, 0.65 mmol) in CH2Cl2 (6.0 mL) were added 6-methoxypyridine-3-carbaldehyde (100 mg, 0.73 mmol), acetic acid (5.0 µL, 87 µmol) and NaBH(OAc)3 (300 mg, 1.4 mmol). After stirring at room temperature overnight, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue (230 mg, 0.40 mmol) in acetone was added p-TsOH·H2O (150 mg, 0.79 mmol). The mixture was concentrated in vacuo. To the residue was added Et2O, and the precipitated solid was collected by filtration to obtain the product (320 mg, 53%). 1H-NMR (DMSO-d6) δ: 1.38–1.65 (2H, m), 1.78–2.17 (3H, m), 2.29 (6H, s), 2.85–3.56 (10H, m), 3.89 (3H, s), 3.99 (2H, d, J = 6.0 Hz), 4.12–4.73 (6H, m), 6.93 (1H, d, J = 8.4 Hz), 7.07–7.16 (4H, m), 7.43–7.60 (5H, m), 7.70 (1H, d, J = 8.6 Hz), 7.75 (2H, d, J = 7.9 Hz), 7.80–7.93 (3H, m), 8.24–8.31 (2H, m), 9.32 (1H, br s), 10.07 (1H, br s); MS m/z: 584 (M + H)+; ESI-MS m/z: 584.2840 (M + H)+ (Calcd for C31H37O3N5F3: 584.2843).
{4-[(4-Chlorophenyl)methyl]piperazin-1-yl}[5-({1-[(6-methoxypyridin-3-yl)methyl]piperidin-4-yl}methoxy)pyridin-2-yl]methanone Ditosylate (15)Compound 15 was prepared from 11 in 72% yield using a similar approach to that described for 14. 1H-NMR (DMSO-d6) δ: 1.39–1.65 (2H, m), 1.81–2.19 (3H, m), 2.29 (6H, s), 2.77–3.60 (10H, m), 3.70–4.74 (11H, m), 6.93 (1H, d, J = 9.0 Hz), 7.12 (4H, dd, J = 0.7, 8.4 Hz), 7.45–7.59 (9H, m), 7.62–7.75 (1H, m), 7.80–7.87 (1H, m), 8.18–8.36 (2H, m), 9.27 (1H, br s), 9.89 (1H, br s); MS m/z: 550 (M + H)+; ESI-MS m/z: 550.2584 (M + H)+ (Calcd for C30H37O3N5Cl: 550.2579).
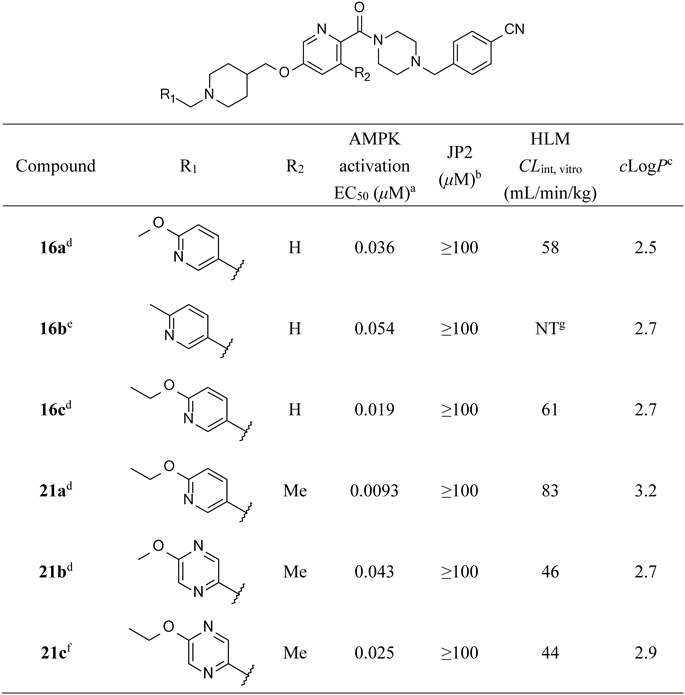
4-({4-[5-({1-[(6-Methoxypyridin-3-yl)methyl]piperidin-4-yl}methoxy)pyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Ditosylate (16a)Compound 16a was prepared from 12 in 66% yield using a similar approach to that described for 14. 1H-NMR (DMSO-d6) δ: 1.35–1.68 (2H, m), 1.78–2.14 (3H, m), 2.29 (6H, s), 2.77–3.60 (10H, m), 3.89 (3H, s), 3.99 (2H, d, J = 6.0 Hz), 4.09–4.71 (6H, m), 6.93 (1H, d, J = 8.4 Hz), 7.12 (4H, d, J = 7.9 Hz), 7.46–7.60 (1H, m), 7.49 (4H, d, J = 7.9 Hz), 7.65–7.76 (3H, m), 7.83 (1H, dd, J = 2.4, 8.6 Hz), 7.97 (2H, d, J = 8.4 Hz), 8.20–8.35 (2H, m), 9.27 (1H, br s), 10.01 (1H, br s); MS m/z: 541 (M + H)+; ESI-MS m/z: 541.2925 (M + H)+ (Calcd for C31H37O3N6: 541.2922).
4-({4-[5-({1-[(6-Methylpyridin-3-yl)methyl]piperidin-4-yl}methoxy)pyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Tetrahydrochloride (16b)Compound 16b was prepared from 12 and 6-methylpyridine-3-carbaldehyde in 38% yield using a similar approach to that described for 13. 1H-NMR (DMSO-d6) δ: 1.54–2.22 (5H, m), 2.57–4.86 (21H, m), 7.54 (1H, dd, J = 2.9, 8.8 Hz), 7.68 (1H, d, J = 8.8 Hz), 7.79–8.04 (4H, m), 8.27 (1H, d, J = 2.9 Hz), 8.38–8.68 (2H, m), 8.86–9.08 (1H, m), 11.34 (1H, br s), 11.91 (1H, br s); MS m/z: 525 (M + H)+; ESI-MS m/z: 525.2971 (M + H)+ (Calcd for C31H37O2N6: 525.2973).
4-({4-[5-({1-[(6-Ethoxypyridin-3-yl)methyl]piperidin-4-yl}methoxy)pyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Ditosylate (16c)Compound 16c was prepared from 12 and 29 in 59% yield using a similar approach to that described for 14. 1H-NMR (DMSO-d6) δ: 1.33 (3H, t, J = 7.1 Hz), 1.41–1.63 (2H, m), 1.76–2.17 (3H, m), 2.29 (6H, s), 2.84–3.54 (10H, m), 3.99 (2H, d, J = 6.0 Hz), 4.12–4.77 (8H, m), 6.89 (1H, d, J = 9.0 Hz), 7.11 (4H, d, J = 8.2 Hz), 7.41–7.59 (1H, m), 7.48 (4H, d, J = 8.2 Hz), 7.65–7.75 (3H, m), 7.82 (1H, dd, J = 2.4, 8.6 Hz), 7.97 (2H, d, J = 8.4 Hz), 8.22–8.34 (2H, m), 9.25 (1H, br s), 10.01 (1H, br s); MS m/z: 555 (M + H)+; ESI-MS m/z: 555.3079 (M + H)+ (Calcd for C32H39O3N6: 555.3078).
Methyl 5-Hydroxy-3-methylpyridine-2-carboxylate (18)To a solution of methyl 5-methoxy-3-methylpyridine-2-carboxylate16) (17; 2.4 g, 13 mmol) in CH2Cl2 (140 mL) was added AlCl3 (20 g, 150 mmol). The mixture was stirred at 55°C overnight under an argon atmosphere. After cooling to 0°C, the mixture was diluted with 1.0 mol/L HCl aqueous solution and stirred at room temperature. The mixture was basified with 1.0 mol/L NaOH aqueous solution and acidified with 10% citric acid aqueous solution. The mixture was extracted with CHCl3/IPA (4 : 1), and the organic layer was dried over MgSO4 and concentrated in vacuo to obtain the product (1.8 g, 80%). 1H-NMR (DMSO-d6) δ: 2.44 (3H, s), 3.79 (3H, s), 7.08 (1H, dd, J = 0.8, 2.6 Hz), 8.02 (1H, dd, J = 0.4, 2.6 Hz), 10.56 (1H, br s); MS m/z: 168 (M + H)+.
5-{[1-(tert-Butoxycarbonyl)piperidin-4-yl]methoxy}-3-methylpyridine-2-carboxylic Acid (19)Compound 19 was prepared from 18 in 69% yield using a similar approach to that described for 9. 1H-NMR (DMSO-d6) δ: 1.03–1.27 (2H, m), 1.40 (9H, s), 1.66–1.82 (2H, m), 1.86–2.06 (1H, m), 2.46–2.54 (3H, m), 2.61–2.92 (2H, m), 3.92–4.03 (2H, m), 3.99 (2H, d, J = 6.4 Hz), 7.36 (1H, d, J = 2.7 Hz), 8.16 (1H, d, J = 2.6 Hz), 12.57 (1H, br s); MS m/z: 351 (M + H)+.
4-[(4-{3-Methyl-5-[(piperidin-4-yl)methoxy]pyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (20)To a solution of 5-{[1-(tert-butoxycarbonyl)piperidin-4-yl]methoxy}-3-methylpyridine-2-carboxylic acid (19; 600 mg, 1.7 mmol), 4-[(piperazin-1-yl)methyl]benzonitrile dihydrochloride (31; 480 mg, 1.8 mmol), Et3N (0.49 mL, 3.5 mmol) in CH2Cl2 (9.0 mL) were added WSC·HCl (450 mg, 2.3 mmol), HOBt (320 mg, 2.4 mmol). After stirring at room temperature overnight, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue in MeOH (5.0 mL) was added 4.0 mol/L HCl in 1,4-dioxane (5.0 mL, 20 mmol). After stirring at room temperature for 1.5 h, the mixture was basified with saturated NaHCO3 aqueous solution and extracted with CHCl3/IPA (4 : 1). The organic layer was dried over MgSO4 and concentrated in vacuo to obtain the product (790 mg, quantitative yield). 1H-NMR (DMSO-d6) δ: 1.07–1.33 (2H, m), 1.62–1.73 (2H, m), 1.75–1.91 (1H, m), 2.21 (3H, s), 2.25–2.37 (2H, m), 2.38–2.55 (4H, m), 2.89–3.04 (2H, m), 3.06–3.16 (2H, m), 3.58–3.61 (2H, m), 3.61–3.69 (2H, m), 3.88 (2H, d, J = 6.6 Hz), 7.32 (1H, d, J = 2.7 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.79 (2H, d, J = 8.4 Hz), 8.06 (1H, d, J = 2.6 Hz); MS m/z: 434 (M + H)+.
4-({4-[5-({1-[(6-Ethoxypyridin-3-yl)methyl]piperidin-4-yl}methoxy)-3-methylpyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Ditosylate (21a)Compound 21a was prepared from 20 and 29 in 56% yield using a similar approach to that described for 14. 1H-NMR (DMSO-d6) δ: 1.33 (3H, t, J = 7.1 Hz), 1.41–1.59 (2H, m), 1.78–2.14 (3H, m), 2.26 (3H, s), 2.29 (6H, s), 2.79–3.78 (10H, m), 3.95 (2H, d, J = 6.0 Hz), 4.06–4.82 (8H, m), 6.90 (1H, d, J = 8.6 Hz), 7.07–7.15 (4H, m), 7.37 (1H, d, J = 2.4 Hz), 7.48 (4H, d, J = 7.9 Hz), 7.61–7.76 (2H, m), 7.82 (1H, dd, J = 2.4, 8.6 Hz), 7.91–8.02 (2H, m), 8.08 (1H, d, J = 2.4 Hz), 8.26 (1H, d, J = 2.4 Hz), 9.27 (1H, br s), 10.04 (1H, br s); MS m/z: 569 (M + H)+; ESI-MS m/z: 569.3238 (M + H)+ (Calcd for C33H41O3N6: 569.3235).
4-({4-[5-({1-[(5-Methoxypyrazin-2-yl)methyl]piperidin-4-yl}methoxy)-3-methylpyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Ditosylate (21b)To a mixture of 4-[(4-{3-methyl-5-[(piperidin-4-yl)methoxy]pyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (20; 410 mg, 0.94 mmol) and MeCN (8.0 mL) were added 2-(chloromethyl)-5-methoxypyrazine (35a; 170 mg, 1.1 mmol) and K2CO3 (390 mg, 2.8 mmol). After stirring at room temperature overnight, the mixture was diluted with H2O and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue (380 mg, 0.68 mmol) in MeOH was added p-TsOH·H2O (240 mg, 1.3 mmol). The mixture was concentrated in vacuo. To the residue was added Et2O, and the precipitated solid was collected by filtration to obtain the product (500 mg, 59%). 1H-NMR (DMSO-d6) δ: 1.42–1.65 (2H, m), 1.76–2.16 (3H, m), 2.26 (3H, s), 2.29 (6H, s), 2.81–3.82 (10H, m), 3.85–4.05 (5H, m), 4.34–4.75 (4H, m), 4.41 (2H, d, J = 4.9 Hz), 7.07–7.15 (4H, m), 7.37 (1H, d, J = 2.7 Hz), 7.48 (4H, d, J = 7.9 Hz), 7.70 (2H, d, J = 8.2 Hz), 7.97 (2H, d, J = 8.2 Hz), 8.09 (1H, d, J = 2.7 Hz), 8.38 (1H, d, J = 1.3 Hz), 8.43 (1H, d, J = 1.3 Hz), 9.66 (1H, br s), 9.78–10.19 (1H, m); MS m/z: 556 (M + H)+; ESI-MS m/z: 556.3030 (M + H)+ (Calcd for C31H38O3N7: 556.3031).
4-({4-[5-({1-[(5-Ethoxypyrazin-2-yl)methyl]piperidin-4-yl}methoxy)-3-methylpyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Succinate (21c)To a solution of 4-[(4-{3-methyl-5-[(piperidin-4-yl)methoxy]pyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (20; 3.2 g, 7.3 mmol), DIPEA (4.0 mL, 23 mmol) and MeCN (30 mL) was added 2-(chloromethyl)-5-ethoxypyrazine (35b; 1.5 g, 8.4 mmol) in MeCN (20 mL). After stirring at room temperature for 14 h, the mixture was concentrated in vacuo. The residue was purified by column chromatography on amino functionalized silica gel (hexane/EtOAc). The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue (3.2 g, 5.7 mmol) in EtOH (12 mL) was added succinic acid (670 mg, 5.7 mmol). After stirring at room temperature for 0.5 h, the mixture was concentrated in vacuo. To the residue was added heptane. After stirring at room temperature overnight, the precipitated solid was collected by filtration to obtain the product (3.7 g, 74%). 1H-NMR (DMSO-d6) δ: 1.19–1.43 (2H, m), 1.34 (3H, t, J = 7.0 Hz), 1.64–1.85 (3H, m), 1.98–2.17 (2H, m), 2.20 (3H, s), 2.25–2.36 (2H, m), 2.39–2.45 (2H, m), 2.41 (4H, s), 2.79–2.96 (2H, m), 3.03–3.21 (2H, m), 3.54–3.69 (6H, m), 3.91 (2H, d, J = 6.0 Hz), 4.33 (2H, q, J = 6.9 Hz), 7.32 (1H, d, J = 2.7 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.79 (2H, d, J = 8.4 Hz), 8.06 (1H, d, J = 2.7 Hz), 8.16 (1H, d, J = 1.3 Hz), 8.21 (1H, d, J = 1.3 Hz), 12.10 (2H, br s); MS m/z: 570 (M + H)+; ESI-MS m/z: 570.3185 (M + H)+ (Calcd for C32H40O3N7: 570.3187).
5-{[1-(tert-Butoxycarbonyl)-4-methoxypiperidin-4-yl]methoxy}-3-methylpyridine-2-carboxylic Acid (22)To a mixture of methyl 5-hydroxy-3-methylpyridine-2-carboxylate (18; 960 mg, 5.7 mmol), tert-butyl 4-(hydroxymethyl)-4-methoxypiperidine-1-carboxylate (37; 1.4 g, 5.7 mmol) and toluene (20 mL) was added cyanomethylenetributylphosphorane (2.2 mL, 8.3 mmol). The mixture was stirred at 100°C overnight under an argon atmosphere. After cooling to room temperature, the mixture was concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/EtOAc). To a solution of the residue in MeOH (20 mL) was added 1.0 mol/L NaOH aqueous solution (20 mL, 20 mmol). The mixture was stirred at 60°C for 1 h. After cooling to room temperature, the mixture was washed with Et2O and the aqueous layer was acidified with 10% citric acid aqueous solution. The mixture was extracted with CHCl3, and the organic layer was dried over MgSO4 and concentrated in vacuo. To the residue was added Et2O, and the precipitated solid was collected by filtration to obtain the product (2.0 g, 91%). 1H-NMR (DMSO-d6) δ: 1.40 (9H, s), 1.45–1.60 (2H, m), 1.73–1.88 (2H, m), 2.51 (3H, s), 2.85–3.16 (2H, m), 3.19 (3H, s), 3.61–3.82 (2H, m), 4.11 (2H, s), 7.42 (1H, d, J = 2.4 Hz), 8.20 (1H, d, J = 2.4 Hz), 12.60 (1H, br s); MS m/z: 381 (M + H)+.
5-{[1-(tert-Butoxycarbonyl)-4-fluoropiperidin-4-yl]methoxy}-3-methylpyridine-2-carboxylic Acid (23)Compound 23 was prepared from 18 and tert-butyl 4-fluoro-4-(hydroxymethyl)piperidine-1-carboxylate18) in 54% yield using a similar approach to that described for 22. 1H-NMR (DMSO-d6) δ: 1.41 (9H, s), 1.55–1.82 (2H, m), 1.83–2.01 (2H, m), 2.51 (3H, s), 2.89–3.18 (2H, m), 3.73–3.93 (2H, m), 4.27 (2H, d, J = 21.2 Hz), 7.42 (1H, d, J = 2.4 Hz), 8.21 (1H, d, J = 2.4 Hz), 12.64 (1H, br s); MS m/z: 369 (M + H)+.
4-[(4-{5-[(4-Methoxypiperidin-4-yl)methoxy]-3-methylpyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (24)Compound 24 was prepared from 22 in quantitative yield using a similar approach to that described for 20. 1H-NMR (DMSO-d6) δ: 1.41–1.62 (2H, m), 1.64–1.83 (2H, m), 2.22 (3H, s), 2.26–2.36 (2H, m), 2.37–2.48 (2H, m), 2.63–2.81 (4H, m), 3.06–3.18 (2H, m), 3.16 (3H, s), 3.60 (2H, s), 3.60–3.70 (2H, m), 4.00 (2H, s), 7.37 (1H, d, J = 2.4 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.80 (2H, d, J = 8.4 Hz), 8.11 (1H, d, J = 2.4 Hz); MS m/z: 464 (M + H)+.
4-[(4-{5-[(4-Fluoropiperidin-4-yl)methoxy]-3-methylpyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (25)Compound 25 was prepared from 23 in quantitative yield using a similar approach to that described for 20. 1H-NMR (DMSO-d6) δ: 1.52–1.90 (4H, m), 2.21 (3H, s), 2.26–2.35 (2H, m), 2.37–2.48 (2H, m), 2.63–2.87 (4H, m), 3.05–3.19 (2H, m), 3.60 (2H, s), 3.61–3.69 (2H, m), 4.14 (2H, d, J = 21.0 Hz), 7.38 (1H, d, J = 2.4 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.79 (2H, d, J = 8.4 Hz), 8.12 (1H, d, J = 2.4 Hz); MS m/z: 452 (M + H)+.
4-({4-[5-({1-[(6-Ethoxypyridin-3-yl)methyl]-4-methoxypiperidin-4-yl}methoxy)-3-methylpyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Succinate (26a)To a solution of 4-[(4-{5-[(4-methoxypiperidin-4-yl)methoxy]-3-methylpyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (24; 370 mg, 0.79 mmol) in CH2Cl2 (6.0 mL) were added 6-ethoxypyridine-3-carbaldehyde (29; 120 mg, 0.79 mmol), acetic acid (5.0 µL, 87 µmol) and NaBH(OAc)3 (450 mg, 2.1 mmol). After stirring at room temperature for 4 h, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue (370 mg, 0.62 mmol) in MeOH was added succinic acid (72 mg, 0.61 mmol). The mixture was concentrated in vacuo. To the residue was added heptane, and the precipitated solid was collected by filtration to obtain the product (340 mg, 61%). 1H-NMR (DMSO-d6) δ: 1.30 (3H, t, J = 7.1 Hz), 1.50–1.70 (2H, m), 1.73–1.87 (2H, m), 2.14–2.63 (8H, m), 2.21 (3H, s), 2.41 (4H, s), 3.05–3.16 (2H, m), 3.15 (3H, s), 3.44 (2H, s), 3.54–3.72 (2H, m), 3.59 (2H, s), 4.00 (2H, s), 4.28 (2H, q, J = 7.1 Hz), 6.75 (1H, d, J = 8.6 Hz), 7.37 (1H, d, J = 2.2 Hz), 7.52 (2H, d, J = 8.2 Hz), 7.61 (1H, dd, J = 2.4, 8.6 Hz), 7.79 (2H, d, J = 8.2 Hz), 8.03 (1H, d, J = 2.2 Hz), 8.10 (1H, d, J = 2.4 Hz), 12.22 (2H, br s); MS m/z: 599 (M + H)+; ESI-MS m/z: 599.3348 (M + H)+ (Calcd for C34H43O4N6: 599.3340).
4-({4-[5-({1-[(6-Ethoxypyridin-3-yl)methyl]-4-fluoropiperidin-4-yl}methoxy)-3-methylpyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Succinate (27a)Compound 27a was prepared from 25 in 68% yield using a similar approach to that described for 26a. 1H-NMR (DMSO-d6) δ: 1.30 (3H, t, J = 7.1 Hz), 1.55–2.06 (4H, m), 2.14–2.48 (6H, m), 2.21 (3H, s), 2.42 (4H, s), 2.58–2.74 (2H, m), 3.03–3.18 (2H, m), 3.46 (2H, s), 3.59 (2H, s), 3.60–3.70 (2H, m), 4.15 (2H, d, J = 20.5 Hz), 4.28 (2H, q, J = 7.1 Hz), 6.75 (1H, d, J = 8.5 Hz), 7.37 (1H, d, J = 2.2 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.62 (1H, dd, J = 2.4, 8.5 Hz), 7.79 (2H, d, J = 8.4 Hz), 8.03 (1H, d, J = 2.2 Hz), 8.11 (1H, d, J = 2.4 Hz), 12.17 (2H, br s); MS m/z: 587 (M + H)+; ESI-MS m/z: 587.3137 (M + H)+ (Calcd for C33H40O3N6F: 587.3140).
4-({4-[5-({1-[(5-Ethoxypyrazin-2-yl)methyl]-4-methoxypiperidin-4-yl}methoxy)-3-methylpyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Succinate (26b)Compound 26b was prepared from 24 and 35b in 65% yield using a similar approach to that described for 21c. 1H-NMR (DMSO-d6) δ: 1.34 (3H, t, J = 7.1 Hz), 1.53–1.72 (2H, m), 1.72–1.88 (2H, m), 2.21 (3H, s), 2.25–2.47 (6H, m), 2.41 (4H, s), 2.53–2.65 (2H, m), 3.05–3.18 (2H, m), 3.15 (3H, s), 3.59 (4H, s), 3.61–3.69 (2H, m), 4.01 (2H, s), 4.33 (2H, q, J = 7.1 Hz), 7.37 (1H, d, J = 2.4 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.79 (2H, d, J = 8.4 Hz), 8.10 (1H, d, J = 2.4 Hz), 8.17 (1H, d, J = 1.3 Hz), 8.21 (1H, d, J = 1.3 Hz), 12.22 (2H, br s); MS m/z: 600 (M + H)+; ESI-MS m/z: 600.3292 (M + H)+ (Calcd for C33H42O4N7: 600.3293).
4-({4-[5-({1-[(5-Ethoxypyrazin-2-yl)methyl]-4-fluoropiperidin-4-yl}methoxy)-3-methylpyridine-2-carbonyl]piperazin-1-yl}methyl)benzonitrile Succinate (27b)To a solution of 4-[(4-{5-[(4-fluoropiperidin-4-yl)methoxy]-3-methylpyridine-2-carbonyl}piperazin-1-yl)methyl]benzonitrile (25; 2.0 g, 4.4 mmol) in MeCN (20 mL) were added DIPEA (2.7 mL, 16 mmol) and 2-(chloromethyl)-5-ethoxypyrazine (35b; 960 mg, 5.5 mmol) in MeCN (5.0 mL). After stirring at 50°C for 18 h, the mixture was concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). The residue was purified by column chromatography on amino functionalized silica gel (hexane/EtOAc). To the residue was added Et2O. After stirring at room temperature overnight, the precipitated solid was collected by filtration. The obtained solid was purified by column chromatography on silica gel (CHCl3/MeOH/EtOAc). To the residue was added Et2O, and the precipitated solid was collected by filtration. To a mixture of the obtained solid (2.0 g, 3.3 mmol), acetone (15 mL) and EtOH (15 mL) was added succinic acid (390 mg, 3.3 mmol). After stirring at room temperature 15 min, to the mixture was added EtOH (15 mL). After stirring at room temperature 0.5 h, the mixture was stirred at 50°C for additional 15 min. The mixture was filtered and the filtrate was concentrated in vacuo. To the residue was added heptane. After stirring at room temperature for 0.5 h, the precipitated solid was collected by filtration to obtain the product (2.1 g, 68%). 1H-NMR (DMSO-d6) δ: 1.34 (3H, t, J = 7.1 Hz), 1.61–2.01 (4H, m), 2.21 (3H, s), 2.26–2.47 (6H, m), 2.42 (4H, s), 2.62–2.81 (2H, m), 3.05–3.19 (2H, m), 3.53–3.72 (2H, m), 3.59 (2H, s), 3.62 (2H, s), 4.16 (2H, d, J = 20.7 Hz), 4.34 (2H, q, J = 7.1 Hz), 7.37 (1H, d, J = 2.4 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.79 (2H, d, J = 8.4 Hz), 8.12 (1H, d, J = 2.4 Hz), 8.18 (1H, d, J = 1.3 Hz), 8.22 (1H, d, J = 1.3 Hz), 12.16 (2H, br s); MS m/z: 588 (M + H)+; ESI-MS m/z: 588.3092 (M + H)+ (Calcd for C32H39O3N7F: 588.3093).
6-Ethoxypyridine-3-carbaldehyde (29)To a solution of 5-bromo-2-ethoxypyridine (28; 5.0 g, 25 mmol) in THF (70 mL) was added dropwise n-BuLi (16 mL, 26 mmol, 1.7 mol/L in hexane) at −68°C under a nitrogen atmosphere. After stirring at the same temperature for 0.5 h, to the mixture was added dropwise DMF (10 mL, 130 mmol). The mixture was allowed to warm gradually to room temperature and stirred for 3 h. The mixture was diluted with 10% citric acid aqueous solution and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/EtOAc) to obtain the product (2.2 g, 59%). 1H-NMR (DMSO-d6) δ: 1.35 (3H, t, J = 7.1 Hz), 4.43 (2H, q, J = 7.1 Hz), 6.96 (1H, ddd, J = 0.7, 0.7, 8.7 Hz), 8.11 (1H, dd, J = 2.4, 8.7 Hz), 8.75 (1H, dd, J = 0.7, 2.4 Hz), 9.96 (1H, d, J = 0.7 Hz); MS m/z: 152 (M + H)+.
4-[(Piperazin-1-yl)methyl]benzonitrile Dihydrochloride (31)To a solution of tert-butyl piperazine-1-carboxylate (30; 7.0 g, 38 mmol) in CH2Cl2 (47 mL) were added 4-formylbenzonitrile (5.9 g, 45 mmol) and acetic acid (4.3 mL, 75 mmol). After stirring at room temperature for 0.5 h, to the mixture was added NaBH(OAc)3 (16 g, 75 mmol). After stirring at room temperature for 3 h, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH). To a solution of the residue in EtOAc (60 mL) and CHCl3 (60 mL) was added 4.0 mol/L HCl in EtOAc (60 mL, 240 mmol). After stirring at room temperature for 3 d, the mixture was diluted with hexane, and then the precipitated solid was collected by filtration to obtain the product (8.0 g, 77%). 1H-NMR (D2O) δ: 3.59 (8H, s), 4.52 (2H, s), 7.70 (2H, d, J = 8.2 Hz), 7.90 (2H, d, J = 8.2 Hz); MS m/z: 202 (M + H)+.
Methyl 5-Methoxypyrazine-2-carboxylate (33a)To a solution of methyl 5-chloropyrazine-2-carboxylate (32; 10 g, 58 mmol) in MeOH (100 mL) was added portion-wise sodium methoxide (5.0 g, 93 mmol) at 0°C under a nitrogen atmosphere. After stirring at room temperature for 3 h, the mixture was diluted with 10% citric acid aqueous solution and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/EtOAc) to give the product (7.9 g, 81%). 1H-NMR (DMSO-d6) δ: 3.88 (3H, s), 4.00 (3H, s), 8.41 (1H, d, J = 1.3 Hz), 8.84 (1H, d, J = 1.3 Hz); MS m/z: 169 (M + H)+.
Ethyl 5-Ethoxypyrazine-2-carboxylate (33b)To a solution of t-BuOK (2.3 g, 21 mmol) in EtOH (30 mL) was added portion-wise methyl 5-chloropyrazine-2-carboxylate (32; 3.0 g, 17 mmol) at 0°C. After stirring at the same temperature for 1 h, the mixture was neutralized with 1.0 mol/L HCl aqueous solution and extracted with CHCl3. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3) to give the product (2.8 g, 82%). 1H-NMR (DMSO-d6) δ: 1.33 (3H, t, J = 7.1 Hz), 1.37 (3H, t, J = 7.1 Hz), 4.34 (2H, q, J = 7.1 Hz), 4.44 (2H, q, J = 7.1 Hz), 8.37 (1H, d, J = 1.3 Hz), 8.81 (1H, d, J = 1.3 Hz); MS m/z: 197 (M + H)+.
(5-Methoxypyrazin-2-yl)methanol (34a)To a mixture of methyl 5-methoxypyrazine-2-carboxylate (33a; 2.3 g, 14 mmol) and MeOH (45 mL) was added NaBH4 (1.6 g, 42 mmol) at 0°C. After stirring at the same temperature for 15 min, the mixture was stirred at room temperature for an additional 2 h. The mixture was acidified with 1.0 mol/L HCl aqueous solution and basified with 1.0 mol/L NaOH aqueous solution. The mixture was extracted with CHCl3/IPA (4 : 1), and the organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/EtOAc) to obtain the product (1.5 g, 77%). 1H-NMR (DMSO-d6) δ: 3.90 (3H, s), 4.55 (2H, d, J = 5.7 Hz), 5.40 (1H, t, J = 5.7 Hz), 8.20–8.22 (1H, m), 8.22–8.24 (1H, m); MS m/z: 141 (M + H)+.
(5-Ethoxypyrazin-2-yl)methanol (34b)Compound 34b was prepared from 33b in 75% yield using a similar approach to that described for 34a. 1H-NMR (DMSO-d6) δ: 1.34 (3H, t, J = 7.1 Hz), 4.34 (2H, q, J = 7.1 Hz), 4.54 (2H, d, J = 5.7 Hz), 5.39 (1H, t, J = 5.7 Hz), 8.07–8.32 (2H, m); MS m/z: 155 (M + H)+.
2-(Chloromethyl)-5-methoxypyrazine (35a)To a solution of (5-methoxypyrazin-2-yl)methanol (34a; 700 mg, 5.0 mmol) in CH2Cl2 (10 mL) was added SOCl2 (1.0 mL, 14 mmol) at 0°C. After stirring at room temperature for 0.5 h, the mixture was concentrated in vacuo to obtain the product (800 mg, quantitative yield). 1H-NMR (CDCl3) δ: 3.98 (3H, s), 4.65 (2H, s), 8.19–8.20 (1H, m), 8.20–8.21 (1H, m); MS m/z: 159 (M + H)+.
2-(Chloromethyl)-5-ethoxypyrazine (35b)Compound 35b was prepared from 34b in 100% yield using a similar approach to that described for 35a. 1H-NMR (DMSO-d6) δ: 1.35 (3H, t, J = 7.1 Hz), 4.36 (2H, q, J = 7.1 Hz), 4.80 (2H, s), 8.29 (1H, d, J = 1.3 Hz), 8.33 (1H, d, J = 1.3 Hz); MS m/z: 173 (M + H)+.
tert-Butyl 4-(Hydroxymethyl)-4-methoxypiperidine-1-carboxylate (37)To a solution of tert-butyl 1-oxa-6-azaspiro[2.5]octane-6-carboxylate19) (36; 10 g, 47 mmol) in MeOH (150 mL) was added p-TsOH·H2O (300 mg, 1.6 mmol). After stirring at room temperature overnight, the mixture was diluted with saturated NaHCO3 aqueous solution and H2O. The mixture was extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/EtOAc) to give the product (6.0 g, 52%). 1H-NMR (DMSO-d6) δ: 1.23–1.43 (2H, m), 1.39 (9H, s), 1.53–1.67 (2H, m), 2.77–3.06 (2H, m), 3.13 (3H, s), 3.30–3.34 (2H, m), 3.53–3.73 (2H, m), 4.54 (1H, t, J = 5.5 Hz); MS m/z: 246 (M + H)+.