Experimental
ChemistryAll of the starting materials were obtained commercially and were used without further purification. All of the reported yields were for isolated products and were not optimized. Melting points were determined in open capillaries on a WRS-1A digital melting point apparatus (Shenguang). 1H-NMR spectra was recorded in dimethyl sulfoxide (DMSO)-d6 on a Bruker DRX-500 (500 MHz) using TMS as internal standard. 13C-NMR spectra was recorded in DMSO-d6 on a Bruker DRX-500 (126 MHz) using TMS as internal standard. The chemical shifts were reported in ppm (δ) and coupling constants (J) values were given in Hertz (Hz). Mass spectra were obtained from Agilent 1100 LC/MSD (Agilent) or Q-tof micro MS (Micromass) and the high-resolution (HR) electrospray ionization-time of flight (ESI-TOF)-MS was recorded on Agilent 6224 A (TOF) LC/MS. The purity of all tested compounds was established by HPLC to be >95.0%. HPLC analysis was performed at room temperature (r.t.) using an Agilent Eclipse XDB-C18 (250 × 4.6 mm) and 30% MeOH/H2O as a mobile phase and plotted at 254 nm.
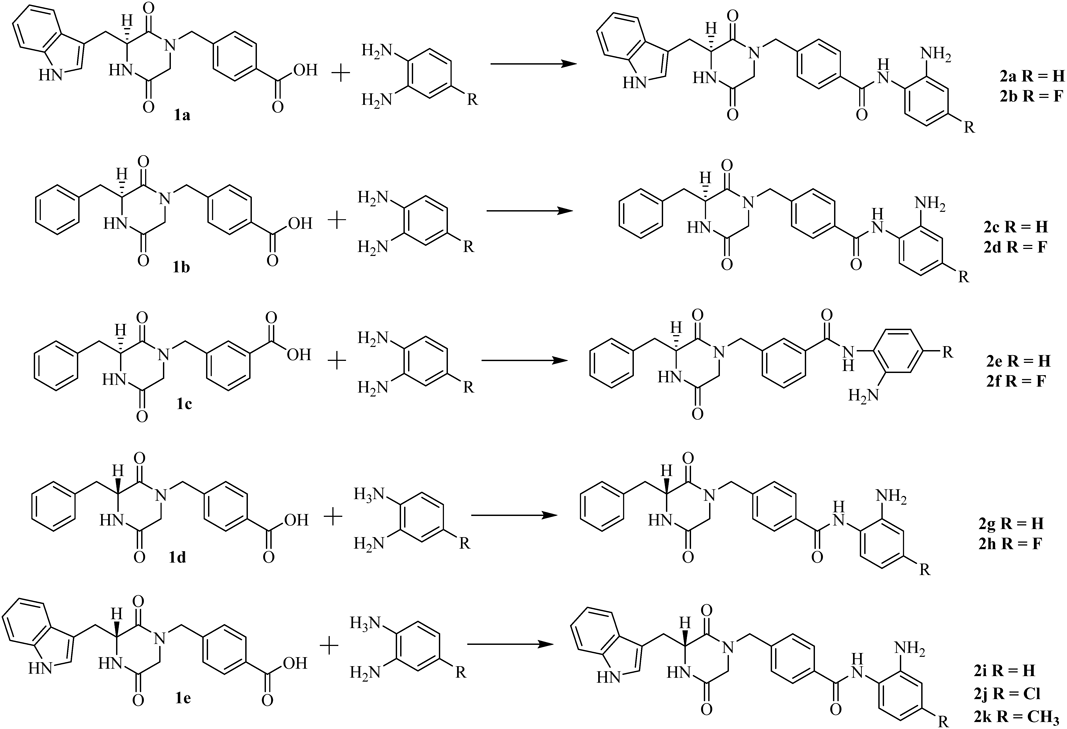
(S)-4-((3-((1H-indol-3-yl)methyl)-2,5-dioxopiperazin-1-yl)methyl)-N-(2-aminophenyl)benzamide (2a)To a stirred mixture of 1a (100 mg, 0.265 mmol), HATU (93.6 mg, 0.292 mmol) and DIPEA (0.184 mL, 1.06 mmol) in 10 mL of DMF was added benzene-1,2-diamine (28.66 mg, 0.265 mmol) at 0°C. After stirring at r.t. for 4 h, the reaction mixture was diluted with saturated sodium chloride (50 mL) and extracted with EtOAc (50 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The product was obtained by chromatography on a silica gel column (70.0 mg, 56.5%). mp: 145–147°C. 1H-NMR (500 MHz, DMSO-d6) δ: 10.98 (s, 1H), 9.60 (s, 1H), 8.38 (s, 1H), 7.82 (d, J = 7.9 Hz, 2H), 7.57 (d, J = 7.9 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.19 (d, J = 7.4 Hz, 1H), 7.12 (t, J = 7.2 Hz, 1H), 7.07 (d, J = 1.7 Hz, 1H), 7.05–6.94 (m, 4H), 6.80 (d, J = 7.2 Hz, 1H), 6.62 (t, J = 7.3 Hz, 1H), 4.92 (s, 2H), 4.35 (dd, J = 34.5, 16.4 Hz, 3H), 3.45–3.38 (m, 2H), 3.08 (dd, J = 14.4, 4.0 Hz, 1H), 2.76 (d, J = 17.1 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.69, 165.60, 165.17, 143.54, 139.73, 136.52, 134.24, 128.45, 127.99, 127.93, 127.09, 126.95, 125.20, 123.84, 121.54, 119.22, 119.03, 116.78, 116.63, 111.82, 108.60, 56.18, 49.01, 48.49, 30.09. HR-MS (ESI, m/z): Calcd for: 468.2030. (C27H26N5O3+, [M + H]+). Found: 468.2033.
(S)-4-((3-((1H-indol-3-yl)methyl)-2,5-dioxopiperazin-1-yl)methyl)-N-(2-amino-4-fluorophenyl) Benza-mide (2b)Using the synthetic method for 2a, compound 1a and 4-fluorobenzene-1,2-diamine gave 2b as a white solid, 61.2% yield. mp: 198–200°C. 1H-NMR (500 MHz, DMSO-d6) δ: 10.99 (s, 1H), 9.54 (s, 1H), 8.39 (s, 1H), 7.82 (d, J = 7.9 Hz, 2H), 7.57 (d, J = 7.9, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.12 (dd, J = 14.8, 7.7 Hz, 2H), 7.07 (d, J = 1.6 Hz, 1H), 7.02 (t, J = 7.7 Hz, 3H), 6.56 (dd, J = 11.2, 2.6 Hz, 1H), 6.45–6.32 (m, 1H), 5.25 (s, 2H), 4.39 (d, J = 15.0 Hz, 1H), 4.31 (d, J = 14.9 Hz, 2H), 4.06 (m, 1H), 3.43–3.38 (m, 1H), 3.08 (dd, J = 14.4, 4.1 Hz, 1H), 2.75 (d, J = 17.1 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.69, 165.85, 165.17, 160.58, 145.90, 139.75, 136.51, 134.10, 128.97, 128.47, 127.97, 127.92, 125.21, 121.54, 119.76, 119.21, 119.02, 111.82, 108.58, 102.53, 101.96, 56.17, 48.98, 48.17, 30.09. HR-MS (ESI, m/z): Calcd for: 486.1936. (C27H25FN5O3+, [M + H]+). Found: 486.1937.
(S)-N-(2-Aminophenyl)-4-((3-benzyl-2,5-dioxopiperazin-1-yl)methyl)benzamide (2c)Using the synthetic method for 2a, compound 1b and benzene-1, 2-diamine gave 2c as a white solid, 75.3% yield. mp: 172–174°C. 1H-NMR (500 MHz, DMSO-d6) δ: 9.70 (s, 1H), 8.40 (s, 1H), 7.96 (d, J = 6.8 Hz, 2H), 7.31–7.18 (m, 6H), 7.14 (d, J = 6.3 Hz, 2H), 7.01 (t, J = 6.9 Hz, 1H), 6.83 (d, J = 7.1 Hz, 1H), 6.64 (t, J = 6.7, 1H), 4.94 (s, 2H), 4.66 (d, J = 14.8 Hz, 1H), 4.33 (d, J = 6.1 Hz, 2H), 3.54 (d, J = 17.2 Hz, 1H), 3.21 (d, J = 13.3 Hz, 1H), 3.06–2.89 (m, 1H), 2.80 (d, J = 17.2 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.04, 165.51, 165.28, 143.66, 139.69, 136.11, 134.32, 130.57, 128.62, 128.45, 127.32, 127.23, 127.01, 123.77, 116.75, 116.61, 56.06, 48.94, 48.67, 39.53. HR-MS (ESI, m/z): Calcd for: 429.1921. (C25H25N4O3+, [M + H]+). Found: 429.1923.
(S)-N-(2-Amino-4-fluorophenyl)-4-((3-benzyl-2,5-dioxopiperazin-1-yl)methyl)benzamide (2d)Using the synthetic method for 2a, compound 1b and 4-fluorobenzene-1,2-diamine gave 2d as a white solid, 66.8% yield. mp: 175–177°C. 1H-NMR (500 MHz, DMSO-d6) δ: 9.62 (s, 1H), 8.40 (s, 1H), 7.95 (d, J = 6.1 Hz, 2H), 7.33–7.19 (m, 5H), 7.15 (s, 3H), 6.58 (d, J = 10.9 Hz, 1H), 6.39 (s, 1H), 5.26 (s, 2H), 4.65 (d, J = 14.7 Hz, 1H), 4.34 (d, J = 12.9 Hz, 2H), 3.53 (d, J = 17.2 Hz, 1H), 3.21 (d, J = 13.1 Hz, 1H), 2.96 (d, J = 13.0 Hz, 1H), 2.80 (d, J = 17.2 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.03, 165.78, 165.27, 160.60, 146.00, 139.72, 136.12, 134.20, 130.56, 129.08, 128.62, 128.48, 128.40, 127.33, 119.72, 102.52, 101.94, 56.06, 48.94, 48.66, 39.53. HR-MS (ESI, m/z): Calcd for: 447.1827. (C25H24FN4O3+, [M + H]+). Found: 447.1828.
(S)-N-(2-Aminophenyl)-3-((3-benzyl-2,5-dioxopiperazin-1-yl)methyl)benzamide (2e)Using the synthetic method for 2a, compound 1c and benzene-1,2-diamine gave 2e as a white solid, 56.3% yield. mp: 174–176°C. 1H-NMR (500 MHz, DMSO-d6) δ: 9.74 (s, 1H), 8.37 (s, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.86 (s, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.34 (d, J = 7.4 Hz, 1H), 7.28–7.12 (m, 4H), 7.09 (d, J = 7.1 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.82 (d, J = 7.8 Hz, 1H), 6.63 (t, J = 7.3 Hz, 1H), 4.93 (s, 2H), 4.66 (d, J = 14.4 Hz, 1H), 4.31 (d, J = 14.1, 2H), 3.53 (d, J = 17.2, 1H), 3.19 (dd, J = 13.4, 3.6 Hz, 1H), 2.94 (d, J = 4.9 Hz, 1H), 2.70 (d, J = 17.3 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.00, 165.70, 165.39, 143.69, 136.50, 135.98, 131.81, 130.53, 128.94, 128.59, 128.54, 127.42, 127.24, 127.21, 127.05, 123.73, 116.76, 116.63, 60.25, 56.02, 48.82, 36.27. HR-MS (ESI, m/z): Calcd for: 429.1921. (C25H25N4O3+, [M + H]+). Found: 429.1923.
(S)-N-(2-amino-4-fluorophenyl)-3-((3-benzyl-2,5-dioxopiperazin-1-yl)methyl)benzamide (2f)Using the synthetic method for 2a, compound 1c and 4-fluorobenzene-1,2-diamine gave 2f as a white solid, 71.6% yield. mp: 123–125°C. 1H-NMR (500 MHz, DMSO-d6) δ: 9.69 (s, 1H), 8.40 (s, 1H), 7.96 (d, J = 6.9 Hz, 2H), 7.35–7.18 (m, 6H), 7.14 (d, J = 6.3 Hz, 2H), 7.01 (t, J = 7.0 Hz, 1H), 6.82 (d, J = 7.6 Hz, 1H), 6.65 (d, J = 6.9 Hz, 1H), 4.93 (s, 2H), 4.66 (d, J = 14.7 Hz, 1H), 4.33 (d, J = 4.4 Hz, 2H), 3.54 (d, J = 17.2 Hz, 1H), 3.28–3.11 (m, 1H), 3.07–2.87 (m, 1H), 2.80 (d, J = 17.2 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.00, 165.39, 162.80, 160.63, 146.00, 136.47, 135.98, 135.28, 131.81, 130.53, 129.05, 128.92, 128.58, 128.54, 127.45, 127.24, 119.68, 102.57, 101.97, 60.25, 56.02, 48.82, 36.27. HR-MS (ESI, m/z): Calcd for: 447.1827. (C25H24FN4O3+, [M + H]+). Found: 447.1828.
(R)-N-(2-Aminophenyl)-4-((3-benzyl-2,5-dioxopiperazin-1-yl)methyl)benzamide (2g)Using the synthetic method for 2a, compound 1d and benzene-1,2-diamine gave 2g as a white solid, 85.7% yield. mp: 168–170°C. 1H-NMR (500 MHz, DMSO-d6) δ: 9.69 (s, 1H), 8.40 (s, 1H), 7.96 (d, J = 6.9 Hz, 2H), 7.35–7.18 (m, 6H), 7.14 (d, J = 6.3 Hz, 2H), 7.01 (t, J = 7.0 Hz, 1H), 6.82 (d, J = 7.6 Hz, 1H), 6.65 (d, J = 6.9 Hz, 1H), 4.93 (s, 2H), 4.66 (d, J = 14.7 Hz, 1H), 4.33 (d, J = 4.4 Hz, 2H), 3.54 (d, J = 17.2 Hz, 1H), 3.28–3.11 (m, 1H), 3.07–2.87 (m, 1H), 2.80 (d, J = 17.2 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.03, 165.51, 165.28, 143.66, 139.69, 136.11, 134.32, 130.57, 128.62, 128.45, 127.32, 127.23, 123.77, 116.74, 116.61, 56.06, 48.94, 48.67, 39.53. HR-MS (ESI, m/z): Calcd for: 429.1921. (C25H25N4O3+, [M + H]+). Found: 429.1923.
(R)-N-(2-Amino-4-fluorophenyl)-4-((3-benzyl-2,5-dioxopiperazin-1-yl)methyl)benzamide (2h)Using the synthetic method for 2a, compound 1d and 4-fluorobenzene-1, 2-diamine gave 2h as a white solid, 77.2% yield. mp: 155–157°C. 1H-NMR (500 MHz, DMSO-d6) δ: 9.69 (s, 1H), 8.40 (s, 1H), 7.96 (d, J = 6.9 Hz, 2H), 7.35–7.18 (m, 6H), 7.14 (d, J = 6.3 Hz, 2H), 7.01 (t, J = 7.0 Hz, 1H), 6.82 (d, J = 7.6 Hz, 1H), 6.65 (d, J = 6.9 Hz, 1H), 4.93 (s, 2H), 4.66 (d, J = 14.7 Hz, 1H), 4.33 (d, J = 4.4 Hz, 2H), 3.54 (d, J = 17.2 Hz, 1H), 3.28–3.11 (m, 1H), 3.07–2.87 (m, 1H), 2.80 (d, J = 17.2 Hz, 1H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.04, 165.78, 165.26, 160.60, 145.98, 139.72, 136.13, 134.20, 130.56, 129.06, 128.61, 128.47, 128.40, 127.32, 119.73, 102.52, 101.94, 56.07, 48.94, 48.67, 39.54. HR-MS (ESI, m/z): Calcd for: 447.1827. (C25H24FN4O3+, [M + H]+). Found: 447.1828.
(R)-4-((3-((1H-Indol-3-yl)methyl)-2,5-dioxopiperazin-1-yl)methyl)-N-(2-aminophenyl)benzamide (2i)Using the synthetic method for 2a, compound 1e and benzene-1, 2-diamine gave 2i as a white solid, 74.6% yield. mp: 161–163°C. 1H-NMR (500 MHz, DMSO-d6) δ: 10.99 (s, 1H), 9.61 (s, 1H), 8.39 (d, 1H, J = 2.3 Hz), 7.82 (d, 2H, J = 8.0 Hz), 7.58 (d, 1H, J = 7.9 Hz), 7.40 (d, 1H, J = 8.1 Hz), 7.19 (d, 1H, J = 7.6 Hz), 7.12 (t, 1H, J = 7.5 Hz), 7.07 (d, 1H, J = 2.0 Hz), 7.01 (m, 4H), 6.81 (d, 1H, J = 7.3 Hz), 6.63 (d, 1H, J = 7.5 Hz), 4.92 (s, 2H), 4.47–4.20 (m, 3H), 3.44–3.37 (m, 2H), 3.08 (dd, 1H, J1 = 14.4 Hz, J2 = 4.3 Hz), 2.76 (d, 1H, J = 17.1 Hz). 13C-NMR (126 MHz, DMSO-d6) δ: 166.65, 165.55, 165.15, 143.58, 139.73, 136.48, 134.20, 128.44, 127.97, 127.90, 127.11, 126.96, 125.20, 123.76, 121.53, 119.21, 119.02, 116.71, 116.57, 111.81, 108.56, 56.15, 48.97, 48.42, 30.07.
(R)-4-((3-((1H-Indol-3-yl)methyl)-2,5-dioxopiperazin-1-yl)methyl)-N-(2-amino-4-chlorophenyl)benzamide (2j)Using the synthetic method for 2a, compound 1e and 4-chlorobenzene-1,2-diamine gave 2i as a white solid, 69.7% yield. mp: 168–170°C. 1H-NMR (500 MHz, DMSO-d6) δ: 10.99 (d, 1H, J = 1.3 Hz), 9.59 (s, 1H), 8.40 (s, 1H), 7.83 (d, 2H, J = 8.0 Hz), 7.59 (d, 1H, J = 7.9 Hz), 7.41 (d, 1H, J = 8.1 Hz), 7.20 (1H, J = 8.4 Hz), 7.12 (t, 1H, J = 7.4 Hz), 7.08 (d, 1H, J = 2.1 Hz), 7.03 (t, 3H, J = 7.8 Hz), 6.85 (d, 1H, J = 2.4 Hz), 6.63 (dd, 1H, J1 = 8.4 Hz, J2 = 2.3 Hz), 5.27 (s, 2H), 4.52–4.21 (m, 3H), 3.44–3.39 (m, 2H), 3.09 (dd, 1H, J1 = 14.4 Hz, J2 = 4.3 Hz), 2.76 (d, 1H, J = 17.1 Hz). 13C-NMR (126 MHz, DMSO-d6) δ: 166.67, 165.78, 165.16, 145.28, 142.41, 139.86, 136.49, 134.02, 130.93, 128.70, 128.51, 127.98, 127.91, 125.21, 122.45, 121.54, 119.22, 119.03, 115.87, 115.25, 111.82, 108.57, 56.16, 48.97, 48.44, 30.09.
(R)-4-((3-((1H-Indol-3-yl)methyl)-2,5-dioxopiperazin-1-yl)methyl)-N-(2-amino-4-methylphenyl)benzamide (2k)Using the synthetic method for 2a, compound 1e and 4-methylbenzene-1,2-diamine gave 2i as a white solid, 55.2% yield. mp: 165–167°C. 1H-NMR (500 MHz, DMSO-d6) δ: 10.98 (s, 1H), 9.53 (s, 1H), 8.38 (d, 1H, J = 2.1 Hz), 7.82 (d, 2H, J = 7.9 Hz), 7.57 (d, 1H, J = 7.9 Hz), 7.40 (d, 1H, J = 8.1 Hz), 7.12 (t, 1H, J = 7.5 Hz), 7.09–7.05 (m, 1H), 7.04 (d, 1H, J = 3.5 Hz), 7.01 (d, 3H, J = 8.2 Hz), 6.62 (s, 1H), 6.44 (d, 1H, J = 7.6 Hz), 4.83 (s, 2H), 4.35 (m, 3H), 3.43–3.37 (m, 2H), 3.08 (dd, 1H, J1 = 14.4 Hz, J2 = 4.3 Hz), 2.76 (d, 1H, J = 17.1 Hz), 2.22 (s, 3H). 13C-NMR (126 MHz, DMSO-d6) δ: 166.65, 165.52, 165.15, 143.38, 139.65, 136.48, 135.97, 134.26, 128.41, 127.96, 127.91, 127.02, 125.20, 121.53, 121.35, 119.21, 119.01, 117.56, 116.97, 111.81, 108.56, 56.15, 48.97, 48.43, 30.07, 21.33.
In Vitro HDAC Enzyme AssayThe inhibitory activity of HDAC isoforms were conducted by the Reaction Biology Corporation, Malvern, PA using HDAC fluorescent activity assay based on the unique Fluor de Lys™ substrate and developer combination. Compounds were dissolved in DMSO and tested in at least 10-dose IC50 mode with 3-fold serial dilution starting at 50 µM. For HDAC assays: Add 2× of HDAC enzyme into reaction plate except control wells (no enzyme), where buffer (50 mM Tris–HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2) was added instead. Add inhibitors in 100% DMSO into the enzyme mixture via acoustic technology (Echo550; nanoliter range). Spin down and preincubate. Add 2× substrate mixture: Fluorogenic HDAC General Substrate: 50 µM, Arg-His-Lys-Lys(Ac). Spin and shake. Incubate for 2 h at 30°C with seal. Add developer with trichostatin A to stop the reaction and to generate fluorescent color. Carry out kinetic measurements for 1.5 h with Envision with 15 min interval (Ex/Em = 360/460 nm). Take end point reading for analysis after the development reaches plateau. GraphPad Prism 5.0 software was used to calculate the IC50 values for each compound.
Cell Culture and Cytotoxicity/Proliferation AssayCulture Medium and Culture Condition of Cell Lines. The cells were cultured in IMDM medium with 20% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin. All cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in air.
Cell Growth Inhibitory Assay. The protocol using Alamar blue reagent for antiproliferative activitives was as follows: 1. plated 100 µL cell suspension or completed medium into 96-well plate using a Matrix 12-channel pipettor. And filled residual wells with 200 µL PBS per well; 2. drugs were added to each well of 96-well plate; 3. placed plate into an incubator with corresponding culture condition for 72 h; 4. pipetted 22 µL Alamar blue solution (1 mM) into each well of 96-well plate; 5. returned plate to incubator and leave for 5–6 h; 6. shook plate for 10 s and recorded fluorescence at 530/590 nm.
Computational MethodsAll computational work was performed in Discovery Studio 3.0 software (BIOVIA, 5005 Wateridge Vista Drive, San Diego, CA92121 U.S.A.). Docking was conducted using cdocker based on the cocrystal of HDAC1 (PDB: 5ICN). HDAC1 was used as the receptor. The cavity occupied by a peptide inhibitor was selected as the ligand binding site. The docking sphere radius value based on the peptide inhibitor was default. Water molecules outside the binding pocket were excluded. The energy minimization for compound 2a, 2c and 2g were performed by Powell’s method for 1000 iterations using Tripos force field and with Gasteiger–Hückel charge. The other docking parameters were kept as default.