Regular Articles
Ring-Opening Cyclization of Spirocyclopropanes Using Sulfoxonium Ylides
2020 Volume 68 Issue 5 Pages 479-486
Details
2020 Volume 68 Issue 5 Pages 479-486
Ring-opening cyclization of cyclohexane-1,3-dione-2-spirocyclopropanes using dimethylsulfoxonium methylide proceeded regioselectively to produce 2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-ones in good to high yields. The reactions of cycloheptane- and cyclopentane-1,3-dione-2-spirocyclopropanes could construct [7.6]- and [5.6]-fused ring systems. This reaction was also carried out using sulfoxonium ethylide, butylide, and benzylide, resulting in the formation of the corresponding 2,3-trans-disubstituted products in good to high yields, and it was shown that the dimethyl group can act as a dummy substituent. It was found that the 2- and 3-phenyhexahydrobenzopyran-5-ones can be readily converted into 5-hydroxyflavan and 5-hydroxyisoflavan, respectively.
In the 1960s, Corey and Chaykovsky developed the reaction of sulfur ylides, such as sulfonium ylide 1 and sulfoxonium ylide 2, with carbonyl compounds, which became known as the Corey–Chaykovsky reaction.1,2) Both 1 and 2 react with either aldehydes or ketones in the same way to give the corresponding epoxides. However, when reacted with enones, they behave differently. For example, dimethylsulfonium methylide (1a) reacts with calcone to afford epoxide in 87% yield, whereas dimethylsulfoxonium methylide (2a) produces cyclopropane in 95% yield3) (Chart 1). Using density functional theory calculations, Yu and colleagues recently reported that these different reaction outcomes can be attributed to thermodynamics, as sulfonium ylide 1a is less stable, and therefore more highly reactive, than sulfoxonium ylide 2a.4) To date, sulfur ylides 1a and 2a have been widely used in the synthesis of various epoxides,5,6) cyclopropanes,7,8) and heterocyclic compounds.9–11)

Doubly-activated cyclopropanes are versatile intermediates for the synthesis of a wide range of carbocycles12) and heterocycles.13) With this in mind, regioselective ring-opening cyclizations of cyclohexane-1,3-dione-2-spirocyclopropanes 3 have been developed for the synthesis of indole14–16) and benzofuran.17,18) Furthermore, we recently reported that the regio- and diastereoselective ring-opening cyclization of spirocyclopropanes 3 with electron-withdrawing group (EWG)-substituted sulfonium ylides 1b affords 2,3-trans-disubstituted 2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-ones 4 in high yields19) (Chart 2A). The reaction proceeds through nucleophilic attack of the carbanion in 1b to the electrophilic cyclopropane carbon possessing an R1 substituent in 3. Subsequently, an SN2-type cyclization occurs to afford the product 4 with the concomitant release of dimethyl sulfide. This was the first example of the ring opening of cyclopropanes using a sulfonium ylide as a carbon nucleophile. However, sulfonium ylides used in this reaction were limited to EWG-stabilized ones 1b. Since sulfur ylides with various reactivities have been used in organic synthesis,5–11) this study focused on the reaction between different sulfur ylides and spirocyclopropanes. Herein, the ring-opening cyclization of spirocyclopropanes 3 using non-, alkyl-, and phenyl-substituted sulfoxonium ylides 2 as a carbon nucleophile is reported (Chart 2B).

Initially, the reaction was carried out using highly reactive dimethylsulfonium methylide (1a), which was prepared in situ from trimethylsulfonium iodide (6) (Chart 3). After treatment of sulfonium salt 6 using sodium hydride in dimethylsulfoxide (DMSO) at room temperature for 0.5 h,3) the produced sulfonium ylide 1a was reacted with 2′-phenylspirocyclopropane 3a.20) Unfortunately, reaction at room temperature for 24 h gave only decomposition products, indicating that the reactivity of 1a is too high to induce a ring-opening cyclization reaction.

Next, the reaction of the lower reactive sulfoxonium ylide 2a with 3a was examined (Table 1). When the dimethylsulfoxonium methylide (2a), which was prepared in situ from 2.1 eq of trimethylsulfoxonium iodide (7a) using 2.0 eq of sodium hydride in DMSO at room temperature for 0.5 h,3) reacted with 3a, the desired ring-opening cyclization proceeded regioselectively at room temperature within 8 h to afford 3-phenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5a) in 68% yield (entry 1). Trimethylsulfoxonium chloride (7b) proved to be a more suitable sulfoxonium ylide precursor than 7a, leading to a higher yield of 5a (76%, entry 2), presumably because side reactions caused by the presence of the highly nucleophilic iodide ion are prevented when switching to the lower nucleophilic chloride ion.18) Changing the solvent from DMSO to N,N-dimethylformamide (DMF) led to a decrease in the product yield to 71% (entry 3). However, neither CH2Cl2 nor toluene21) were found to be suitable solvents, leading to the recovery of 3a (entries 4 and 5). In contrast, when tetrahydrofuran (THF) was used as a solvent (at reflux for 3 h), the reaction with 3a gave the product 5a in 75% yield (entry 6). Changing the base from sodium hydride to potassium tert-butoxide22) (tBuOK in THF at reflux for 3 h) led to a similar product yield of 5a in both cases (74%, entry 7). For ylide preparation in THF, the reaction required 3 h at reflux, whereas in DMSO it required only 0.5 h at room temperature. For this reason, DMSO was chosen as a solvent. Next, an attempt was made to reduce the amount of 7b and base used (entries 8–10), where 1.5 eq of 7b and 1.2 eq of sodium hydride were found to give the best result. Moreover, increasing the reaction temperature to 50°C led to a lower product yield (72%, entry 11). Finally, 1.5 eq of 7b and 1.2 eq of sodium hydride in DMSO at room temperature were chosen as the optimized conditions.
 | |||||
|---|---|---|---|---|---|
| Entry | 7 (eq) | Base (eq) | Solvent | Time (h) | Yield (%) |
| 1 | 7a (2.1) | NaH (2.0) | DMSO | 8 | 68 |
| 2 | 7b (2.1) | NaH (2.0) | DMSO | 8 | 76 |
| 3 | 7b (2.1) | NaH (2.0) | DMF | 8 | 71 |
| 4b) | 7b (2.1) | NaH (2.0) | CH2Cl2 | 24 | NR |
| 5b) | 7b (2.1) | NaH (2.0) | Toluene | 24 | NR |
| 6b) | 7b (2.1) | NaH (2.0) | THF | 8 | 75 |
| 7b) | 7b (2.1) | tBuOK (2.0) | THF | 8 | 74 |
| 8 | 7b (2.1) | NaH (1.5) | DMSO | 12 | 79 |
| 9 | 7b (1.5) | NaH (1.2) | DMSO | 12 | 82 |
| 10 | 7b (1.2) | NaH (1.1) | DMSO | 12 | 68 |
| 11c) | 7b (1.5) | NaH (1.2) | DMSO | 12 | 72 |
a) All reactions were performed on a 0.3 mmol scale. b) Sulfoxonium ylide 2a was prepared by heating at reflux for 3 h. c) The reaction was carried out at 50°C.
Next, the reactions of a range of spirocyclopropanes 3b–i, 8, and 10 with sulfoxonium salt 7b were investigated (Table 2). Under the previously stated optimized conditions, dimedone-derived spirocyclopropane 3b gave the corresponding hexahydrobenzopyran-5-one 5b in 83% yield (entry 1). The reaction of aryl-substituted spirocyclopropanes 3c and 3d bearing p-methyl or p-bromo substituents on their benzene rings afforded products 5c and 5d in 82 and 71% yields, respectively (entries 2 and 3). It was also possible to use 2′,3′-nonsubstituted spirocyclopropanes 3e–h23) in this reaction, furnishing the corresponding products 5e–h in 71–78% yields (entries 4–7). The reaction of n-butyl-substituted spirocyclopropane 3i was then investigated (entry 8). Unexpectedly, the 4-butyl-substituted product 5i′ was obtained together with 3-butyl-substituted 5i, where the combined yield of these products was 83% and the ratio of 5i to 5i′ was determined to be 76 : 24 from 1H-NMR analysis of the crude product. Since the butyl-substituted carbocation, formally formed by the cleavage of cyclopropane, is less stable than phenyl-substituted one, it is speculated that the sulfoxonium ylide 2a would partially attack to the less hindered carbon atom on cyclopropane in 3i to produce the regioisomer 5i′ as a minor product. Next, the reactions of spirocyclopropanes 8 and 10 derived from cycloheptane- and cyclopentane-1,3-diones were investigated (entries 9 and 10). Although the yields of the corresponding products 9 and 11 decreased to 64 and 41%, respectively, it was discovered that the present reaction can also be used to construct [7.6]- and [5.6]-fused ring systems.
 |
a) All reactions were performed on a 0.3 mmol scale. After preparation of sulfoxonium ylide 2a from 1.5 eq of sulfoxonium salt 7b with 1.2 eq of sodium hydride in DMSO for 0.5 h in situ, spirocyclopropane 3, 8 or 10 was added to the reaction mixture. b) The ratio of 5i to 5i′ was determined from 1H-NMR analysis of the crude product.
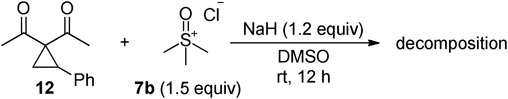
In addition, the reaction of non-spirocyclopropane was examined under the optimized conditions (Chart 4). The reaction of 1,1-diacetyl-2-phenylcyclopropane (12) with sulfoxonium salt 7b in the presence of sodium hydride in DMSO at room temperature for 12 h resulted in the formation of only decomposition products, suggesting that a spiro-type structure is crucial for successful ring-opening cyclization to occur. It is clear that the spirocyclopropane is more reactive than non-spiro one, probably due to its higher ring strain energy.
As an extension of the present method, the reaction was carried out using sulfoxonium ethylide, butylide, and benzylide. Initially, the corresponding sulfoxonium salts were prepared as sulfoxonium ylide precursors (Chart 5). Since the Shea group has already proven that triethylsulfoxonium chloride (14) is a precursor of sulfoxonium ethylide 2b,24) salt 14 was prepared from diethyl sulfide (13) according to their reported procedure. Ethylation of 13 with iodoethane and iodine, anion exchange with benzyltributylammonium chloride, and oxidation using m-chloroperoxybenzoic acid (mCPBA) resulted in the formation of sulfoxonium salt 14 (Chart 5a). Next, tributylsulfoxonium chloride (17) was prepared according to the same procedure. Alkylation of dibutyl sulfide (15) followed by an anion exchange reaction gave tributylsulfonium chloride (16). However, the oxidation of 16 with mCPBA was not successful. As a result of the testing of different oxidation conditions, it was found that the Sharpless oxidation (cat. RuCl3, NaIO4)25) could be applied to this step, resulting in the formation of sulfoxonium salt 17 in 16% yield from 15 (Chart 5b). Because of the difficulty in synthesizing the tribenzylsulfoxonium salt, benzyldimethylsulfoxonium chloride (20) was successfully prepared as an alternative. Although the sulfoxonium ylide precursor 20 has the potential to form methylide in addition to benzylide, it was expected that the deprotonation of 20 would proceed at the most basic benzylic position. Alkylation of dimethyl sulfide (18) with benzyl bromide and a subsequent anion exchange reaction gave benzyldimethylsulfonium chloride (19). Finally, the Sharpless oxidation of 19 provided the corresponding sulfoxonium salt 20 in 44% yield from benzyl bromide (Chart 5c).

Using these sulfoxonium ylide precursors, the reactions of spirocyclopropanes with sulfoxonium ethylide 2b, butylide 2c, and benzylide 2d were investigated (Chart 6). After treatment of triethylsulfoxonium chloride (14) under the aforementioned optimized conditions, spirocyclopropane 3a was added. The reaction proceeded at room temperature within 12 h, affording 2-methyl-3-phenylhexahydrobenzopyran-5-one (5j) in 73% yield (Chart 6a). From this result, it was found that sulfoxonium ethylide 2b was generated from 14 under these conditions.26) The stereochemistry of 5j was determined from 1H nuclear Overhauser effect experiments to be 2,3-trans (see Supplementary materials), as in previous results.19) Tributylsulfoxonium chloride (17) was determined to be a very good precursor in the formation of sulfoxonium butylide 2c to furnish the final trans-3-phenyl-2-propyl-substituted product 5k in 72% yield (Chart 6b). Next, the reaction of 3a with benzyldimethylsulfoxonium chloride (20) was examined under the same conditions, which was found to proceed smoothly to afford trans-2,3-diphenyl-substituted 5l as the sole product in 90% yield (Chart 6c). This result clearly implies that the corresponding sulfoxonium benzylide 2d was formed as expected. The reaction of 2′,3′-nonsubstituted spirocyclopropane 3e with 20 proceeded at 50°C, providing the 2-phenyl-substituted product 5k27,28) in 84% yield (Chart 6c). In the case of the reactions of 14 and 17, either diethyl sulfoxide or dibutyl sulfoxide are expelled as by-products in the cyclization step. However, the reaction of 20 produces dimethyl sulfoxide as a leaving group. It is very interesting and useful in terms of atom economy that the dimethyl group in 20 can act as a dummy substituent.

To demonstrate the utility of this reaction, the conversions of hexahydrobenzopyran-5-ones 5a and 5m into isoflavan and flavan derivatives 21 and 22 were carried out (Chart 7), because isoflavan and flavan are structural motifs found in a number of biologically active natural products.29–32) On the basis of a reported procedure for the aromatization of cyclohexenone,33) reaction of 5a and 5m with iodine in methanol followed by aromatization using a combination of LiI and Li2CO3 afforded 5-hydroxyisoflavan (21) and 5-hydroxyflavan (22) in 68 and 61% yields, respectively.

In conclusion, a regioselective ring-opening cyclization of spirocyclopropanes with dimethylsulfoxonium methylide has been developed, which affords 2-nonsubstituted hexahydrobenzopyran-5-ones in up to 83% yields. Spirocyclopropanes derived from cycloheptane- and cyclopentane-1,3-diones could be applied to this reaction. It was also found that sulfoxonium ethylide, butylide, and benzylide can be used in the present protocol. Moreover, it was shown that the dimethyl group can act as a dummy substituent. The obtained products were readily converted into flavan and isoflavan derivatives. Further application of the present method in the synthesis of a variety of flavan- and isoflavan-based natural products is currently in progress.
Melting points are uncorrected. IR spectra were recorded on a JASCO FT/IR-460 Plus spectrophotometer and absorbance bands are reported in wavenumber (cm−1). All NMR spectra were recorded using a JEOL JNM-ECX400P spectrometer. 1H-NMR spectra were recorded at 400 or 500 MHz. Chemical shifts are reported relative to internal standard (tetramethylsilane at δH 0.00, CDCl3 at δH 7.26, or (CD3)2SO at δH 2.50). Data are presented as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet), coupling constant and integration. 13C-NMR spectra were recorded at 100 or 126 MHz. The following internal reference was used (CDCl3 at δC 77.0 or (CD3)2SO at δC 39.5). All 13C-NMR spectra were determined with complete proton decoupling. High-resolution mass spectra (HR-MS) were determined with JEOL JMS-GCmate II instrument [electron ionization (EI)] and Thermo Scientific LTQ Orbitrap XL ETD [electrospray ionization (ESI)]. Column chromatography was performed on Silica Gel 60 PF254 (Nacalai Tesque, Kyoto, Japan) and Kanto silica gel 60 N (63–210 mesh) under pressure. Analytical TLC was carried out on Merck Kieselgel 60 F254 plates. Visualization was accomplished with UV light and phosphomolybdic acid stain solution followed by heating.
Reagents were used as received unless otherwise noted. Sodium hydride (60% dispersion in mineral oil) was washed with three portions of dry hexane to remove the mineral oil and the remaining sodium hydride was dried in vacuo. Dehydrated DMSO, DMF, CH2Cl2, toluene, THF, acetonitrile, and methanol were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan) and Wako Pure Chemical Corporation (Osaka, Japan). 1-Phenylspiro[2.5]octane-4,8-dione (3a),20) 6,6-dimethyl-1-phenylspiro[2.5]octane-4,8-dione (3b),20) 1-(4-methylphenyl)spiro[2.5]octane-4,8-dione (3c),20) 1-(4-bromophenyl)spiro[2.5]octane-4,8-dione (3d),20) spiro[2.5]octane-4,8-dione (3e),23) 6,6-dimethylspiro[2.5]octane-4,8-dione (3f),23) 6-methylspiro[2.5]octane-4,8-dione (3g),23) 6-phenylspiro[2.5]octane-4,8-dione (3h),23) 1-butylspiro[2.5]octane-4,8-dione (3i),17) 1-phenylspiro[2.4]heptane-4,7-dione (10),20) 1,1-diacetyl-2-phenylcyclopropane (12),23) and triethylsulfoxonium chloride (14)24) were prepared according to literature procedures.
Typical Procedure for the Ring-Opening Cyclization of Spirocyclopropane 3 with Sulfoxonium Salt 7: 3-Phenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5a) (Table 1, Entry 9)Trimethylsulfoxonium chloride (7b) (58 mg, 0.45 mmol) was added to a suspension of sodium hydride (8.6 mg, 0.36 mmol) in DMSO (1.5 mL) at room temperature. After stirring for 30 min, 1-phenylspiro[2.5]octane-4,8-dione (3a) (64 mg, 0.30 mmol) was added to the mixture. After stirring at room temperature for 12 h, the reaction was quenched with water (3 mL) and the whole mixture was extracted with EtOAc (3 × 10 mL). The combined organic layer was washed with brine (10 mL) and dried over anhydrous MgSO4. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, 30% EtOAc in hexane) to provide 5a (56 mg, 82%) as a white solid: mp 117.0–117.5°C; IR (KBr, cm−1) 2949, 1640, 1617, 1398, 1379, 1215, 1188, 1129, 1008, 981, 764, 702; 1H-NMR (400 MHz, CDCl3) δ: 7.34 (t, J = 7.2 Hz, 2H), 7.26 (t, J = 7.2 Hz, 1H), 7.21 (d, J = 7.2 Hz, 2H), 4.32 (ddd, J = 10.8, 4.0, 2.0 Hz, 1H), 3.90 (td, J = 10.8, 2.8 Hz, 1H), 3.04 (tt, J = 10.8, 4.0 Hz, 1H), 2.73 (ddd, J = 16.4, 4.0, 2.0 Hz, 1H), 2.46–2.29 (m, 5H), 2.04–1.92 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 198.1, 171.0, 140.6 128.7, 127.3, 127.1, 111.6, 71.4, 37.8, 36.6, 28.4, 24.9, 20.8; HR-MS (EI) m/z Calcd for C15H16O2 (M+) 228.1150. Found 228.1142.
7,7-Dimethyl-3-phenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5b) (Table 2, Entry 1)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5b was prepared from 3b (73 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 25% EtOAc in hexane) to provide 5b (64 mg, 83%) as a white solid: mp 92.0–92.5°C; IR (KBr, cm−1) ν 2952, 1651, 1624, 1455, 1395, 1377, 1215, 1168, 1125, 1026, 765, 702; 1H-NMR (400 MHz, CDCl3) δ: 7.34 (t, J = 7.6 Hz, 2H), 7.26 (t, J = 7.6 Hz, 1H), 7.21 (d, J = 7.6 Hz, 2H), 4.31 (ddd, J = 10.4, 4.0, 2.4 Hz, 1H), 3.92 (t, J = 10.4 Hz, 1H), 3.04 (tt, J = 10.4, 4.0 Hz, 1H), 2.74 (m, 1H), 2.38–2.22 (m, 5H), 1.09 (s, 3H), 1.08 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 197.7, 169.2, 140.6, 128.7, 127.3, 127.1, 110.2, 71.4, 50.5, 42.1, 37.7, 32.1, 29.0, 27.7, 24.5; HR-MS (EI) m/z Calcd for C17H20O2 (M+) 256.1463. Found 256.1450.
3-(4-Methylphenyl)-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5c) (Table 2, Entry 2)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5c was prepared from 3c (68 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 30% EtOAc in hexane) to provide 5c (60 mg, 82%) as a white solid: mp 72.5–73.0°C; IR (KBr, cm−1) ν 2940, 1645, 1620, 1518, 1463, 1395, 1213, 1186, 1128, 1080, 1009, 980, 817; 1H-NMR (400 MHz, CDCl3) δ: 7.14 (d, J = 8.0 Hz, 2H), 7.10 (d, J = 8.0 Hz, 2H), 4.29 (ddd, J = 10.8, 4.0, 2.4 Hz, 1H), 3.86 (t, J = 10.8 Hz, 1H), 3.00 (tt, J = 10.8, 4.0 Hz, 1H), 2.71 (ddq, J = 16.4, 5.6, 1.6 Hz, 1H), 2.45–2.38 (m, 4H), 2.33 (s, 3H), 2.31 (m, 1H), 2.05–1.91 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 198.0, 170.9, 137.4, 136.7, 129.4, 127.1, 111.6, 71.4, 37.3, 36.6, 28.3, 24.8, 21.0, 20.8; HR-MS (EI) m/z Calcd for C16H18O2 (M+) 242.1307. Found 242.1340.
3-(4-Bromophenyl)-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5d) (Table 2, Entry 3)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5d was prepared from 3d (88 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 40% EtOAc in hexane) to provide 5d (65 mg, 71%) as a white solid: mp 125.0–125.5°C; IR (KBr, cm−1) ν 2939, 1645, 1619, 1489, 1462, 1395, 1214, 1187, 1130, 1106, 1072, 1008, 978, 825; 1H-NMR (400 MHz, CDCl3) δ: 7.46 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 4.28 (ddd, J = 10.4, 3.2, 2.4 Hz, 1H), 3.87 (t, J = 10.4 Hz, 1H), 3.01 (tt, J = 10.4, 3.2 Hz, 1H), 2.71 (dd, J = 16.4, 3.2 Hz, 1H), 2.45–2.35 (m, 4H), 2.29 (dd, J = 16.4, 10.4 Hz, 1H), 2.04–1.93 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 197.9, 170.9, 139.5, 131.8, 129.0, 120.9, 111.3, 71.0, 37.2, 36.6, 28.3, 24.7, 20.8; HR-MS (EI) m/z Calcd for C15H15BrO2 (M+) 306.0255. Found 306.0235.
2,3,4,6,7,8-Hexahydro-5H-1-benzopyran-5-one (5e) (Table 2, Entry 4)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5e was prepared from 3e (41 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 40% EtOAc in hexane) to provide 5e (35 mg, 78%) as a white solid: mp 41.5–42.0°C; IR (KBr, cm−1) ν 2941, 1644, 1617, 1399, 1275, 1228, 1178, 1131, 1092, 964, 859; 1H-NMR (400 MHz, CDCl3) δ: 4.10 (t, J = 5.2 Hz, 2H), 2.39–2.35 (m, 4H), 2.24 (t, J = 6.4 Hz, 2H), 1.95 (quint, J = 6.4 Hz, 2H), 1.90–1.84 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 198.2, 171.5, 111.6, 67.3, 36.6, 28.6, 21.4, 20.8, 17.5; HR-MS (EI) m/z Calcd for C9H12O2 (M+) 152.0837. Found 152.0801.
7,7-Dimethyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5f) (Table 2, Entry 5)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5f was prepared from 3f (50 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 25% EtOAc in hexane) to provide 5f (40 mg, 74%) as a colorless oil: IR (film, cm−1) ν 2955, 1654, 1626, 1397, 1372, 1270, 1232, 1200, 1165, 1125, 1089, 1065, 960; 1H-NMR (500 MHz, CDCl3) δ: 4.10 (t, J = 5.0 Hz, 2H), 2.26–2.23 (m, 6H), 1.87 (quint, J = 5.0 Hz, 2H), 1.06 (s, 6H); 13C-NMR (126 MHz, CDCl3) δ: 197.9, 169.7, 110.2, 67.4, 50.5, 42.4, 31.9, 28.5, 28.3, 21.4, 17.2; HR-MS (EI) m/z Calcd for C11H16O2 (M+) 180.1150. Found 180.1146.
7-Methyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5g) (Table 2, Entry 6)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5g was prepared from 3g (46 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 40% EtOAc in hexane) to provide 5g (37 mg, 74%) as a white solid: mp 76.0–76.5°C; IR (KBr, cm−1) ν 2948, 1644, 1615, 1396, 1298, 1225, 1134, 1066, 964, 916; 1H-NMR (400 MHz, CDCl3) δ: 4.17 (dt, J = 10.8, 6.4 Hz, 1H), 4.02 (ddd, J = 10.8, 7.6, 4.0 Hz, 1H), 2.44 (m, 1H), 2.36 (dd, J = 15.6, 2.4 Hz, 1H), 2.31–2.01 (m, 5H), 1.93–1.81 (m, 2H), 1.06 (d, J = 5.6 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ: 198.2, 170.9, 111.1, 67.4, 45.0, 36.7, 28.4, 21.4, 21.0, 17.4; HR-MS (EI) m/z Calcd for C10H14O2 (M+) 166.0994. Found 166.0998.
7-Phenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5h) (Table 2, Entry 7)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5h was prepared from 3h (64 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 30% EtOAc in hexane) to provide 5h (48 mg, 71%) as a white solid: mp 78.0–78.5°C; IR (KBr, cm−1) ν 2944, 1647, 1613, 1398, 1276, 1182, 1128, 1091, 1062, 964, 763, 704; 1H-NMR (400 MHz, CDCl3) δ: 7.34 (t, J = 7.2 Hz, 2H), 7.25–7.23 (m, 3H), 4.22 (m, 1H), 4.05 (td, J = 10.4, 3.6 Hz, 1H), 3.34 (tt, J = 11.6, 4.4 Hz, 1H), 2.71–2.54 (m, 4H), 2.36–2.21 (m, 2H), 1.91–1.90 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 197.2, 170.6, 143.0, 128.7, 126.9, 126.6, 111.4, 67.6, 43.7, 38.9, 36.1, 21.4, 17.5; HR-MS (EI) m/z Calcd for C15H16O2 (M+) 228.1150. Found 228.1147.
3-Butyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5i) and 4-Butyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5i′) (Table 2, Entry 8)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5i and 5i′ were prepared from 3i (58 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 15% EtOAc in hexane) to provide 5i (38 mg, 62%) as a colorless oil and 5i′ (13 mg, 21%) as a colorless oil. 5i: IR (neat, cm−1) ν 2929, 1655, 1625, 1395, 1377, 1215, 1188, 1132, 1006, 754; 1H-NMR (400 MHz, CDCl3) δ: 4.15 (ddd, J = 10.4, 3.6, 2.4 Hz, 1H), 3.59 (t, J = 10.4 Hz, 1H), 2.49 (ddd, J = 15.2, 4.4, 2.0 Hz, 1H), 2.42–2.30 (m, 4H), 2.01–1.89 (m, 2H), 1.87–1.70 (m, 2H), 1.46–1.19 (m, 6H), 0.90 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ: 198.3, 171.3, 111.1, 71.5, 36.6, 31.5, 31.4, 28.7, 28.4, 24.1, 22.7, 20.8, 13.9; HR-MS (EI) m/z Calcd for C13H20O2 (M+) 208.1463. Found 208.1445. 5i′: IR (neat, cm−1) ν 2952, 1653, 1615, 1396, 1372, 1277, 1182, 1124, 1083, 1022; 1H-NMR (400 MHz, CDCl3) δ: 4.18 (dtd, J = 10.8, 4.0, 1.2 Hz, 1H), 4.03 (td, J = 10.8, 4.0 Hz, 1H), 2.61 (m, 1H), 2.43–2.27 (m, 4H), 1.92 (quint, J = 6.4 Hz, 2H), 1.81–1.70 (m, 2H), 1.61 (m, 1H), 1.38–1.23 (m, 4H), 1.11 (tdd, J = 9.2, 8.4, 5.2 Hz, 1H), 0.89 (t, J = 6.4 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ: 197.7, 170.7, 116.1, 63.5, 37.2, 33.5, 29.4, 28.8, 26.7, 24.7, 22.7, 20.9, 14.1; HR-MS (EI) m/z Calcd for C13H20O2 (M+) 208.1463. Found 208.1483.
Preparation of 1-Phenylspiro[2.6]nonane-4,9-dione (8) (Table 2, Entry 9)According to our reported procedure for the synthesis of spirocyclopropanes from cycloalkane-1,3-diones with sulfonium salt,20) 8 was prepared from cycloheptane-1,3-dione with (2-bromo-1-phenylethyl)dimethylsulfonium bromide. Powdered K2CO3 (726 mg, 3.00 mmol) and cycloheptane-1,3-dione (221 mg, 1.75 mmol) were added to a suspension of the sulfonium salt (857 mg, 2.63 mmol) in EtOAc (17.5 mL). After stirring at room temperature for 8 h, the sulfonium salt (286 mg, 0.87 mmol) and powdered K2CO3 (242 mg, 1.75 mmol) were further added to the reaction mixture. After stirring at room temperature for 2 h, the reaction mixture was filtered through a Celite pad and the filter cake was rinsed with EtOAc (100 mL). Combined filtrates were washed with water (30 mL) and the aqueous layer was extracted with EtOAc (2 × 15 mL). The combined organic layer was washed with brine (30 mL) and dried over anhydrous MgSO4. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, 1 : 1 Et2O/hexane) to provide 8 (332 mg, 83%) as a white solid: mp 76.5–77.0°C; IR (KBr, cm−1) ν 2945, 1701, 1677, 1496, 1457, 1333, 1221, 1205, 1127, 1110, 966, 948, 897, 782, 754; 1H-NMR (400 MHz, CDCl3) δ: 7.28–7.19 (m, 3H), 7.12–7.09 (m, 2H), 3.37 (t, J = 8.0 Hz, 1H), 2.73 (m, 1H), 2.64 (dt, J = 12.8, 4.4 Hz, 1H), 2.39 (dd, J = 8.0, 4.0 Hz, 1H), 2.32 (m, 1H), 2.03–1.86 (m, 4H), 1.81 (m, 1H), 1.71 (dd, J = 8.8, 4.4 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ: 206.0, 205.3, 133.9, 128.3, 128.2, 127.4, 52.4, 41.9, 41.8, 38.7, 24.0, 22.3, 20.8; HR-MS (EI) m/z Calcd for C15H16O2 (M+) 228.1150. Found 228.1163.
3-Phenyl-3,4,6,7,8,9-hexahydrocyclohepta[b]pyran-5(2H)-one (9) (Table 2, Entry 9)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 9 was prepared from 8 (68 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 20% EtOAc in hexane) to provide 9 (47 mg, 64%) as a colorless oil: IR (film, cm−1) ν 2939, 1639, 1611, 1455, 1375, 1349, 1235, 1195, 1182, 1152, 1107, 1029, 995, 956, 757, 700; 1H-NMR (400 MHz, CDCl3) δ: 7.33 (t, J = 7.2 Hz, 2H), 7.27–7.19 (m, 3H), 4.28 (dt, J = 10.4, 4.0 Hz, 1H), 3.84 (t, J = 10.4 Hz, 1H), 2.96 (tt, J = 10.4, 4.0 Hz, 1H), 2.81 (dd, J = 16.4, 4.0 Hz, 1H), 1.80 (m, 1H), 2.63–2.59 (m, 3H), 2.32 (dd, J = 16.8, 10.4 Hz, 1H), 1.92–1.75 (m, 4H); 13C-NMR (100 MHz, CDCl3) δ: 201.3, 171.4, 140.9, 128.7, 127.3, 127.0, 114.0, 71.3, 41.2, 37.7, 32.1, 27.5, 23.5, 21.1; HR-MS (EI) m/z Calcd for C16H18O2 (M+) 242.1307. Found 242.1320.
3-Phenyl-3,4,6,7-tetrahydrocyclopenta[b]pyran-5(2H)-one (11) (Table 2, Entry 10)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 11 was prepared from 10 (60 mg, 0.30 mmol) with 7b (58 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 40% EtOAc in hexane) to provide 11 (26 mg, 41%) as a white solid: mp 107.5–108.0°C; IR (KBr, cm−1) ν 2932, 1681, 1625, 1460, 1402, 1237, 1116, 956, 768, 708; 1H-NMR (400 MHz, CDCl3) δ: 7.36 (t, J = 7.2 Hz, 2H), 7.28 (t, J = 7.2 Hz, 1H), 7.21 (d, J = 7.2 Hz, 2H), 4.44 (ddd, J = 10.8, 4.0, 2.0 Hz, 1H), 4.09 (t, J = 10.8 Hz, 1H), 3.06 (tt, J = 10.8, 4.4 Hz, 1H), 2.63–2.55 (m, 3H), 2.51–2.48 (m, 2H), 2.36 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ: 203.5, 183.9, 139.8, 128.9, 127.4, 115.3, 73.3, 37.5, 33.7, 26.2, 23.4; HR-MS (EI) m/z Calcd for C14H14O2 (M+) 214.0994. Found 214.1007.
Preparation of Tributylsulfoxonium Chloride (17) (Chart 5)Dibutyl sulfide (15) (920 mg, 5.00 mmol), butyl iodide (805 mg, 5.50 mmol) and iodine (635 mg, 2.50 mmol) were heated at 70°C and stirred in the dark for 24 h. The resulting mixture was then transferred to a flask containing water (15 mL) and CH2Cl2 (10 mL). After the addition of benzyltributylammonium chloride (1.56 g, 5.00 mmol), the reaction mixture was stirred at room temperature for 24 h. The aqueous layer was separated from the reaction mixture and washed with CH2Cl2 (5 × 10 mL). Evaporation of water in vacuo furnished the crude product 16 (1.05 g, approx. 4.40 mmol) as a colorless oil, which was used in the next step without further purification.
RuCl3 (46 mg, 0.22 mmol) was added to a biphasic solution of crude product 16 in acetonitrile (6.3 mL), CH2Cl2 (6.3 mL), and water (9.4 mL). The mixture was stirred vigorously at room temperature for 10 min and then NaIO4 (3.77 g, 17.6 mmol) was added in 5 portions over 10 min to the brown colored solution. After stirring at room temperature for 12 h, the reaction was quenched with MeOH (20 mL) and the resulting suspension was filtered through a Celite pad, and the filter cake was rinsed with MeOH (20 mL). The filtrate was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, 4 : 4 : 1 CH2Cl2/hexane/MeOH) to provide 17 (206 mg, 18% from 15) as a white solid: mp 99.0–99.5°C; IR (KBr, cm−1) ν 2960, 2872, 1469, 1385, 1183, 1091, 936, 750; 1H-NMR (400 MHz, CDCl3) δ: 4.39 (t, J = 7.6 Hz, 6H), 1.93 (quint, J = 7.6 Hz, 6H), 1.60 (sext, J = 7.6 Hz, 6H), 1.03 (t, J = 7.6 Hz, 9H); 13C-NMR (100 MHz, CDCl3) δ: 49.6, 22.0, 21.6, 13.5; HR-MS (ESI+) m/z Calcd for C24H54ClO2S2 [2(nBu3SO)+Cl−]+ 473.3248. Found 473.3261.
Preparation of Benzyldimethylsulfoxonium Chloride (20) (Chart 5)Dimethyl sulfide (18) (1.86 g, 30.0 mmol) and benzyl bromide (855 mg, 5.00 mmol) were stirred at room temperature for 24 h. The resulting white precipitate was collected by suction, washed with Et2O (10 mL), dried in vacuo, and then dissolved in water (15 mL) and CH2Cl2 (10 mL). After the addition of benzyltributylammonium chloride (1.56 g, 5.00 mmol), the reaction mixture was stirred at room temperature for 24 h. The aqueous layer was separated from the reaction mixture and washed with CH2Cl2 (5 × 10 mL). Evaporation of water in vacuo furnished the crude product 19 (630 mg, approx. 3.34 mmol) as a colorless oil, which was used in the next step without further purification.
RuCl3 (35 mg, 0.17 mmol) was added to a biphasic solution of crude product 19 in acetonitrile (4.8 mL), CH2Cl2 (4.8 mL), and water (7.1 mL). The mixture was stirred vigorously at room temperature for 10 min and then NaIO4 (2.86 g, 13.4 mmol) was added in 5 portions over 10 min to the brown colored solution. After stirring at room temperature for 12 h, the reaction was quenched with MeOH (20 mL) and the resulting suspension was filtered through a Celite pad, and the filter cake was rinsed with MeOH (20 mL). The filtrate was concentrated in vacuo and the crude solid was purified by recrystallization from MeOH/CHCl3 to provide 20 (446 mg, 44% from benzyl bromide) as a white solid: mp 162.5–163.0°C; IR (KBr, cm−1) ν 2962, 2878, 1496, 1236, 1050, 958, 776, 700; 1H-NMR (400 MHz, (CD3)2SO) δ: 7.56–7.50 (m, 5H), 5.76 (s, 2H), 3.91 (s, 6H); 13C-NMR (100 MHz, (CD3)2SO) δ: 131.8, 130.3, 129.4, 123.8, 56.5, 36.4; HR-MS (EI) m/z Calcd for C9H13ClOS (M+) 204.0376. Found 204.0390.
rel-(2R,3S)-2-Methyl-3-phenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5j) (Chart 6)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5j was prepared from 3a (64 mg, 0.30 mmol) with 1424) (77 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 40% EtOAc in hexane) to provide 5j (53 mg, 73%) as a white solid: mp 131.5–132.0°C; IR (KBr, cm−1) ν 2930, 1646, 1616, 1455, 1392, 1378, 1244, 1227, 1188, 1100, 1030, 987, 865, 758, 702; 1H-NMR (400 MHz, CDCl3) δ: 7.31 (t, J = 7.2 Hz, 2H), 7.24 (t, J = 7.2 Hz, 1H), 7.13 (d, J = 7.2 Hz, 2H), 4.12 (dq, J = 10.0, 6.4 Hz, 1H), 2.68 (dd, J = 16.0, 5.2 Hz, 1H), 2.60 (td, J = 10.0, 5.2 Hz, 1H), 2.45–2.37 (m, 4H), 2.31 (dd, J = 16.0, 11.2 Hz, 1H), 2.06–1.92 (m, 2H), 1.14 (d, J = 6.4 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ: 198.0, 171.0, 141.7, 128.7, 127.7, 127.0, 111.9, 77.7, 45.2, 36.6, 28.5, 26.8, 20.9, 19.2; HR-MS (EI) m/z Calcd for C16H18O2 (M+) 242.1307. Found 242.1303.
rel-(2R,3S)-2-Propyl-3-phenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5k) (Chart 6)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5k was prepared from 3a (64 mg, 0.30 mmol) with 17 (115 mg, 0.45 mmol). The crude product was purified by column chromatography (silica gel, 30% EtOAc in hexane) to provide 5k (58 mg, 74%) as a colorless oil: IR (neat, cm−1) ν 2957, 1652, 1624, 1392, 1223, 1187, 1112, 959, 760, 702; 1H-NMR (400 MHz, CDCl3) δ: 7.31 (t, J = 7.2 Hz, 2H), 7.24 (tt, J = 7.2, 1.6 Hz, 1H), 7.12 (dd, J = 7.2, 1.6 Hz, 2H), 4.03 (ddd, J = 8.8, 7.6, 4.0 Hz, 1H), 2.72–2.65 (m, 2H), 2.45–2.37 (m, 4H), 2.30 (ddt, J = 17.2, 12.0, 2.0 Hz, 1H), 2.04–1.93 (m, 2H), 1.51 (m, 1H), 1.41–1.26 (m, 3H), 0.82 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ: 198.0, 171.1, 142.0, 128.7, 127.7, 126.8, 111.7, 80.9, 43.3, 36.6, 34.8, 28.5, 27.0, 20.9, 18.1, 13.8; HR-MS (EI) m/z Calcd for C18H22O2 (M+) 270.1620. Found 270.1620.
rel-(2R,3S)-2,3-Diphenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5l) (Chart 6)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5l was prepared from 3a (43 mg, 0.20 mmol) with 20 (61 mg, 0.30 mmol). The crude product was purified by column chromatography (silica gel, 40% EtOAc in hexane) to provide 5l (55 mg, 90%) as a white solid: mp 127.0–127.5°C; IR (KBr, cm−1) ν 2931, 1645, 1617, 1455, 1391, 1340, 1227, 1189, 1128, 1092, 1008, 982, 764; 1H-NMR (400 MHz, CDCl3) δ: 7.19–7.07 (m, 8H), 6.95 (d, J = 6.8 Hz, 2H), 4.94 (d, J = 10.0 Hz, 1H), 3.05 (dt, J = 10.0, 5.2 Hz, 1H), 2.79 (dd, J = 16.8, 5.2 Hz, 1H), 2.59–2.38 (m, 5H), 2.04 (quint, J = 6.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ: 198.0, 171.0, 140.4, 138.3, 128.3, 128.1, 128.0, 127.9, 127.0, 126.7, 112.0, 83.8, 45.2, 36.6, 28.5, 26.3, 20.9; HR-MS (EI) m/z Calcd for C21H20O2 (M+) 304.1463. Found 304.1488.
2-Phenyl-2,3,4,6,7,8-hexahydro-5H-1-benzopyran-5-one (5m)27,28) (Chart 6)According to the typical procedure for the ring-opening cyclization of 3a with 7b, 5m was prepared from 3e (41 mg, 0.30 mmol) with 20 (92 mg, 0.45 mmol) at 50°C. The crude product was purified by column chromatography (silica gel, 8 : 1.5 : 0.5 CH2Cl2/hexane/EtOAc) to provide 5m (57 mg, 84%) as a colorless oil: IR (neat, cm−1) ν 2943, 1651, 1622, 1394, 1293, 1181, 1073, 759, 700; 1H-NMR (400 MHz, CDCl3) δ: 7.41–7.32 (m, 5H), 4.94 (dd, J = 10.4, 2.4 Hz, 1H), 2.52–2.35 (m, 5H), 2.28 (m, 1H), 2.17 (m, 1H), 2.05–1.97 (m, 2H) 1.90 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ: 198.2, 171.5, 140.3, 128.6, 128.2, 125.9, 111.6, 79.1, 36.7, 29.1, 28.7, 20.9, 18.1.
Typical Procedure for the Synthesis of Isoflavan and Flavan Derivatives: 5-Hydroxy-3-phenyl-3,4-dihydro-2H-1-benzopyran (21) (Chart 7)Iodine (228 mg, 0.90 mmol,) was added to a solution of 5a (68 mg, 0.30 mmol) in MeOH (1.5 mL) at room temperature. After stirring for 3 h, the reaction mixture was quenched by addition of saturated aqueous Na2S2O3 (3 mL), and the resulting mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water (10 mL) and brine (10 mL), and dried over anhydrous MgSO4. Filtration and evaporation in vacuo furnished the crude product (152 mg), which was used in the next step without further purification.
LiI (24 mg, 0.33 mmol) and Li2CO3 (44 mg, 0.33 mmol) were added to a solution of crude product in DMF (3.0 mL). After stirring at reflux for 2.0 h, the reaction was cooled to room temperature and diluted with 20% EtOAc in hexane (3 mL). The reaction mixture was quenched by addition of saturated aqueous NH4Cl (3 mL), and the resulting mixture was extracted with 20% EtOAc in hexane (3 × 10 mL). The combined organic layers were washed with water (10 mL) and brine (10 mL), and dried over anhydrous MgSO4. Filtration was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, 40% EtOAc in hexane) to provide 21 (46 mg, 68%) as a pale yellow solid: mp 79.5–80.0°C; IR (KBr, cm−1) ν 3400, 2925, 1707, 1616, 1595, 1496, 1470, 1460, 1329, 1276, 1234, 1091, 1073, 1029, 980, 774, 757, 700; 1H-NMR (400 MHz, CDCl3) δ: 7.36 (t, J = 7.6 Hz, 2H), 7.29–7.24 (m, 3H), 6.99 (t, J = 8.4 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 6.37 (d, J = 8.4 Hz, 1H), 4.87 (s, 1H), 4.35 (dq, J = 10.4, 1.6 Hz, 1H), 4.00 (t, J = 10.4, 1H), 3.22 (tt, J = 10.8, 4.0 Hz, 1H), 3.07 (ddd, J = 16.4, 5.6, 1.6 Hz, 1H), 2.80 (dd, J = 16.4, 12.4 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ: 155.5, 154.0, 141.3, 128.8, 127.4, 127.2, 127.1, 109.7, 109.1, 106.7, 70.4, 38.1, 26.6; HR-MS (EI) m/z Calcd for C15H14O2 (M+) 226.0994. Found 226.0999.
5-Hydroxy-2-phenyl-3,4-dihydro-2H-1-benzopyran (22) (Chart 7)According to the typical procedure for the synthesis of 21, 22 was prepared from 5m (57 mg, 0.25 mmol). The crude product was purified by column chromatography (silica gel, 20% EtOAc in hexane) to provide 22 (34 mg, 61%) as a pale yellow solid: mp 103.5–104.0°C; IR (KBr, cm−1) ν 3404, 2925, 1704, 1616, 1592, 1496, 1465, 1339, 1278, 1201, 1055, 1017, 774, 758, 700; 1H-NMR (400 MHz, CDCl3) δ: 7.44–7.37 (m, 4H), 7.32 (t, J = 7.2 Hz, 1H), 6.99 (t, J = 8.0 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 6.37 (d, J = 8.0 Hz, 1H), 5.01 (dd, J = 10.4, 2.4 Hz, 1H), 4.77 (s, 1H), 2.86–2.72 (m, 2H), 2.25 (m, 1H), 2.07 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ: 156.3, 153.9, 141.5, 128.5, 127.9, 127.2, 126.0, 109.6, 109.4, 106.6, 77.4, 29.3, 19.4; HR-MS (EI) m/z Calcd for C15H14O2 (M+) 226.0994. Found 226.1004.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.