Abstract
A unique phenomenon in solid tumors, the enhanced permeability and retention (EPR) effect is now well known in the development of macromolecular anticancer therapy. However, cancers with low vascular permeability have posed a challenge for these EPR based therapeutic systems. An intrinsic vascular modulator, such as nitric oxide (NO), could augment the endogenous EPR effect. However, the most important aim has been to construct an effective NO delivery system for cancer. Since it is well known that human serum albumin is one of the most important endogenous NO transport proteins in human circulation, for more than a decade we have demonstrated that S-nitrosated human serum albumin dimer (SNO-HSA-Dimer) becomes an enhancer of the EPR effect. Here, we summarize the enhanced effect of SNO-HSA-Dimer on the anticancer effect of macromolecular anticancer drugs such as PEGylated liposomal doxorubicin (Doxil®). In C26-bearing mice with highly permeable vasculature, SNO-HSA-Dimer is able to increase more 3-fold the tumor accumulation of these anticancer drugs, thereby tripling their anticancer effects. Interestingly, the tumor accumulation of Doxil® in B16-bearing mice, which are characterized by a low permeable vasculature, increased more than 6-fold in the presence of SNO-HSA-Dimer, and the improved accumulation of Doxil® led to both increased survival and decreased tumor volume. These results strongly suggest that the more cancer is refractory, the more the SNO-HSA-Dimer could enhance the EPR effect via an endogenous albumin transport (EAT) system. Accordingly, we conclude that the EAT system is promising as a drug delivery system (DDS) strategy for refractory cancer therapy.
1. Introduction
In the cancer microenvironment observed in refractory pancreatic cancer, many cell types, including cancer cells, immune cells and vascular endothelial cells, are irregular, and hypoxia and undernutrition brings diversity to the phenotypes of these cells. It is extremely difficult to deliver an anticancer agent to such a “chaotic” cancer microenvironment. So far, “active” or/and “passive” targeting has been attempted in the tumor targeting of anticancer drugs in cancer chemotherapy. “Active” targeting results in an increase in the cell population in which the target does not act, because the target is expressed only in limited cells. On the other hand, “passive” targeting can be applied to a relatively wide range of cancer types, due to the enhanced permeability and retention (EPR) effect that is the basis of its theory. However, with passive targeting, clinical data indicate that tumor accumulation is only approximately 10% of the dose, and satisfactory results are only rarely obtained using solely an EPR effect strategy. Therefore, many questions the existence of an EPR effect itself in the treatment of human cancer.1,2) Therefore, the question of whether it is possible to efficiently deliver a high-molecular weight anticancer drug to the entire tumor is an urgent issue, with a strong clinical demand; it is a critical for drug delivery system (DDS) researchers to contribute to resolving this. In this review, we focus on the high tumor accumulation of albumin, which has been demonstrated through the strategy of enhancing the EPR effect using the S-nitrosated human serum albumin dimer (SNO-HSA Dimer). We would like to also introduce a DDS strategy utilizing the endogenous albumin transport (EAT) system to tumor cells, as well as its potential for future development.
2. Human Serum Albumin and EPR Effect
Human serum albumin (HSA) is the most abundant protein in human serum. HSA possesses transport activities of exogenous and endogenous ligands, as well as antioxidative and enzymatic actions1) (Fig. 1). HSA also has a very long half-life (approximately 20 d in humans), which is an excellent advantage in using HSA as a biomimetic DDS carrier.3–8) In fact, conjugation technology with HSA has been used to improve the retention of molecules and peptides with a short half-life in blood, which has improved the development of therapeutic drugs for cancer, hepatitis, diabetes, and rheumatism.9–14)
Regarding DDS in cancer treatment, the accumulation of Evans blue-conjugated albumin in tumor tissue, which led to the discovery of the EPR effect, would be an advantage of using HSA as a DDS carrier for anticancer drugs.15–23) The EPR effect, first reported by Maeda and Matsumura in 1986, was well-known to be induced by a marked increase in vascular permeability in tumor tissues, in addition to a lack of development of the lymphatic system. In this first report of the EPR effect, they mentioned that endogenous high molecular weight proteins (>50 kDa), such as serum proteins albumin, are affected by the EPR effect.15) This means that molecules that have a high affinity for HSA, such as Evans blue, will bind to endogenous serum proteins immediately after administration, and are affected by the EPR effect. These results strongly indicate that HSA and antibodies also accumulate in tumor tissues via the EPR effect.
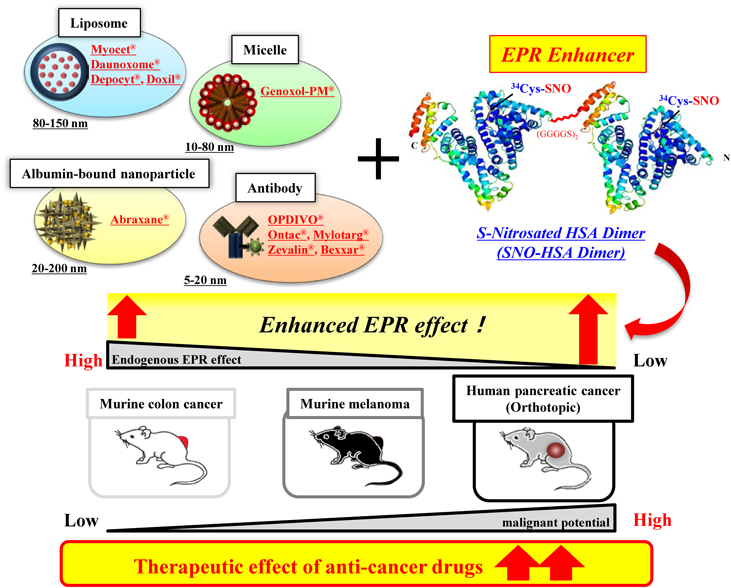
3. Development of SNO-HSA Dimer as an EPR Enhancer
The EPR effect has become an essential theory in the development of nanomedicines in current cancer treatments, and Doxil®, one of the nanomedicines, is being developed and approved in many countries. Although survival rates and the reduction of adverse effects are improved by nanomedicines, some studies have shown that EPR effect-based medicines were ineffective or poorly effective depending on differences in cancer type or tumor size.1,2) In addition, it is difficult to evaluate the heterogeneity of the EPR effect using a uniform tumor-bearing animal model. We have also experienced an extremely large difference in endogenous EPR effect between mouse colon cancer-derived Colon26 cells and mouse melanoma B16 cells.24) Therefore, it is essential to reconstruct an experimental system with the consideration of the type and size of the tumor. Hence, we propose a next-generation EPR treatment strategy by not only “utilizing” but actually “controlling” the intrinsic EPR effect. The merit of this approach is that the simple co-administration of macromolecular anticancer drugs with an EPR enhancer could increase the tumor accumulation of macromolecular drugs. The EPR enhancer could reduce adverse effects and increase the potential maximum dose of macromolecular anticancer drugs. It is expected that the clinical utility of this EPR enhancer strategy can be relatively easily improved, and that its versatility will be high. On the other hand, an EPR enhancer strategy may also enhance the supply of oxygen and nutrients, which may increase tumor growth. Therefore, the method of EPR enhancer strategy should be transient.
Thus, nitric oxide (NO), which is a vasodilator in vivo, was selected as an EPR enhancer; S-nitrosated human serum albumin dimer (SNO-HSA-Dimer) was developed. HSA Dimer has a beneficial characteristic of prolonged blood half-life and increased accumulation in tumor tissue compared with the HSA monomer. The SNO-HSA Dimer obtained by adding NO (which has an EPR controlling activity) to the HSA Dimer, which is a tumor selective carrier, is highly likely to be an attractive EPR enhancer agent. By means of HSA Dimer production using recombinant DNA technology, uniform high-purity proteins can be obtained without the molecular heterogeneity found in micelles and liposomes. Our previous papers indicated that SNO-HSA Dimer has been able to transiently enhance the EPR effect (up to 3 h immediately after administration) by delivering NO in a tumor tissue-specific manner, and then returning to its previous state.24,25) This ‘reversible’ EPR enhancement phenomenon is crucial in enhancing EPR without contributing to unexpected tumor growth, and this result strongly suggests that SNO-HSA Dimer is not only an EPR ‘enhancing’ agent but also a ‘controlling’ agent. Next, the usefulness of this SNO-HSA Dimer as an EPR regulator will be mainly described with reference to examples verified in vivo.
4. Application of EPR Enhancer by SNO-HSA Dimer
First, we used Doxil®, a doxorubicin-encapsulated polyethylene glycol (PEG) liposome, as a macromolecular anticancer drug, and examined its combined effect with SNO-HSA Dimer on the antitumor effect of Doxil®. The two tumor-bearing mice models we used were Colon 26 (C26) cells derived from mouse colon cancer having a large endogenous EPR effect, and mouse melanoma cells B16 cells having a small endogenous EPR effect. In each mouse model, it was previously confirmed that the administration of SNO-HSA Dimer alone did not inhibit tumor growth. Since Doxil® has a passive targeting effect via the EPR effect, Doxil® alone was expected to possess an anti-tumor effect in mouse models that have a large endogenous EPR effect, such as C26 cell tumor-bearing mice. Thus, the antitumor effect of Doxil® alone was observed in the C26 cell tumor-bearing model, but not in the B16 cell tumor-bearing model (Fig. 2). Intriguingly, when SNO-HSA Dimer was co-administered to each tumor-bearing mice model (C26 cell and B16 cell), both models showed significantly higher antitumor effects than when Doxil® alone was administered. The SNO-HSA Dimer also enhanced the antitumor effect of Doxil® in a B16 cell tumor-bearing model in which Doxil® alone was ineffective (Fig. 2). To confirm that this effect was based on an improvement in the amount of Doxil® in the tumor, the amount of doxorubicin in the tumor was quantified. These results showed that, with the addition of the SNO-HSA Dimer, the amounts of doxorubicin in the tumor were more than three times and six times higher in the C26 (Table 1) and B16 cell tumor-bearing models (Table 2), respectively. On the other hand, the distribution of doxorubicin in other organs, such as the liver and kidney, were reduced compared to the Doxil® alone groups. We also confirmed that the combined administration of SNO-HSA Dimer prolonged the survival rate, and suppressed lung metastasis, in both C26 cell and B16 cell tumor-bearing models. These results indicate that SNO-HSA Dimer actively transports macromolecular anticancer drugs, even in situations where Doxil® has extremely low tumor accumulation (the endogenous EPR effect is small).
Table 1. Quantification of Doxorubicin in Tissues of C26 Cell Tumor-bearing Model
| Doxil® | Doxil® + SNO-HSA Dimer | p Value |
|---|
| Kidney | 2.2 ± 2.1 | N.D. | p < 0.01 |
| Liver | 4.5 ± 2.1 | 2.2 ± 2.1 | p < 0.05 |
| Plasma | 27.2 ± 5.7 | 19.2 ± 7.8 | |
| Tumor | 10.1 ± 2.3 | 34.2 ± 5.1 | p < 0.01 |
Tumors reaching approximately 100 mm3 were selected for the study. Selected mice were randomized into 3 groups of 3–4 mice per group for the following i.v. treatments; Doxil® (6.0 mg (DOX)/kg), Doxil® combined with SNO-HSA-Dimer (1.3 µmol NO/kg), and no treatment (control group). At 24 h after injection, the mice were sacrificed, and tumors, liver and kidneys were collected. Dox distribution (ng/mg protein or mg/mL plasma), N.D. stands for Not Detectable.
Table 2. Quantification of Doxorubicin in Tissues of B16 Cell Tumor-bearing Model
| Doxil® | Doxil® + SNO-HSA Dimer | p Value |
|---|
| Kidney | 32.2 ± 5.6 | 19.7 ± 4.5 | p < 0.01 |
| Liver | 20.3 ± 2.9 | 12.4 ± 3.1 | p < 0.05 |
| Plasma | 26.6 ± 7.7 | 14.7 ± 5.8 | |
| Tumor | 4.7 ± 2.3 | 30.2 ± 3.1 | p < 0.01 |
Tumors reaching approximately 100 mm3 were selected for the study. Selected mice were randomized into 3 groups of 3–4 mice per group for the following i.v. treatments; Doxil® (6.0 mg (DOX)/kg), Doxil® combined with SNO-HSA-Dimer (1.3 µmol NO/kg), and no treatment (control group). At 24 h after injection, the mice were sacrificed, and tumors, liver and kidneys were collected. Dox distribution (ng/mg protein or mg/mL plasma), N.D. stands for Not Detectable.
Here, we have introduced the example of the SNO-HSA enhanced application of Doxil®, but it has also been confirmed that the antitumor effect of micelle and albumin nanoparticle formulations can be similarly enhanced by SNO-HSA Dimer co-administration24,26) (Fig. 3). Recently, similar studies regarding EPR enhancers have reported. It has been observed that the administration of NO-encapsulated liposomes, or even NO itself, could enhance the EPR effect, but these enhanced ratios were minimal compared with the results using SNO-HSA Dimer.27,28)
5. The Effect of Tumor Accumulation of Human Serum Albumin beyond the EPR Effect
In general, there are three steps involved in the delivery of anticancer drugs to tumor tissues: from the whole body to the tumor stroma (STEP 1), from the tumor stroma to the tumor cells (STEP 2), and finally, uptake by the tumor cells (STEP 3) (Fig. 4). As shown in the figure, the EPR effect contributes to STEP 1 alone among these three steps, and it is considered that a strategy based solely on EPR effect is insufficient for targeting anticancer drugs to tumor cells. Therefore, the most important thing is to add a driving force for delivering a drug into tumor cells, in addition to the EPR effect.
Recently, it has been reported that HSA possesses a targeting ability beyond the EPR effect. The enhanced uptake of albumin-based DDS in solid tumors is mediated by the pathophysiology of tumor tissue. The accumulation of HSA is also due to the transcytosis initiated by the binding of albumin to 60-kDa glycoprotein (gp60),29–33) as well as to secreted protein acid rich in cysteine (SPARC),34–38) which is found in many types of tumor stroma39) (Fig. 4). This evidence indicates that HSA itself is not only “passively” but also “actively” targeted to tumor cells, and among the three steps, it can break through STEP 2 and STEP 3. Why do tumor cells actively uptake HSA? One likely answer is that tumor cells have a survival mechanism that actively takes up HSA as a “nutritional” source. In particular, this phenomenon is remarkably observed in pancreatic ductal adenocarcinoma with poor angiogenesis, in which it has been reported that HSA uptaken by cancer cells via micropinocytosis is reused as an amino acid source.40) We propose naming this phenomenon the “EAT System.” Tumor cells actively “EAT” endogenous HSA as a source of amino acids to continue cell proliferation. In a region around a blood vessel with flow, HSA is transported along with the blood flow. Therefore, the EAT System is not required for tumor tissues rich in blood vessels. On the other hand, in a hypoxic region where blood vessels are poor, the EAT System should be activated to survive. Interestingly, the expression of SPARC increases in response to hypoxia.41) Taken together, the region in which the EAT System is activated is considered to be a region where it is traditionally difficult to deliver a polymer anticancer agent, that is, a region where the EPR effect must be enhanced. Therefore, it is likely that HSA is the best carrier to deliver the EPR enhancer NO to tumor tissues.
6. Prospects for the Future
Here, a strategy for controlling the EPR effect has been described using the SNO-HSA Dimer employing the EAT System. Our preliminary data suggests that the strategy for controlling the EPR may apply not only to liposome preparations, such as Doxil®, and albumin-binding drugs such as Abraxane®, but also to micelle preparations and antibodies such as OPDIVO®, which has recently received prominent attention. We believe that this EPR control technology will contribute not only to reducing the mental, physical and economic burden on patients by reducing the administration period and dosage, but also to medical economics in general. The potential widespread use of EPR control is extremely high and is considered significant.
In addition, it has been confirmed that this approach to EPR controlling using SNO-HSA Dimer is more effective in tumors with low endogenous EPR effect and low blood vessel density (such as melanoma and pancreatic cancer). This amazing result could be achieved with only the SNO-HSA Dimer, which has a high ability to deliver tumors via the EAT System. Based on the above, we plan to develop HSA nanoparticles using the EAT System, combining EPR regulators with anticancer agents, diagnostic agents, etc. Further, gp60 in vascular endothelial cells and SPARC in tumor stroma are involved in albumin tumor transport.42,43) It has recently been reported that caveolin-1-mediated macropinocytosis is critical for the tumor cell uptake of Abraxane®.44) Hence, the screening of compounds that increase the expression of gp60, SPARC, and caveolin-1, or the development of HSA mutants that increase an affinity for gp60, SPARC, and caveolin-1, are among the strategies for improving the EAT System. Some research and development underway to determine polymer anticancer agents in which a plurality of drugs capable of performing multi-drug chemotherapy in a single drug is encapsulated in liposomes or micelles. The hope is that multiple drug-binding sites of HSA could be used to easily develop multi-drug-conjugated albumin nanoparticles.
7. Conclusion
More than 30 years after the discovery of the EPR effect, a number of macromolecular drugs have been researched, developed, and clinically applied based on this theory. As shown by the huge numbers of paper citations, this discovery is truly an original finding; such impactful discoveries are rare. However, the accumulation of multifaceted data, including clinical data, has cast doubt on the universality of the EPR effect, and DDS researchers are at crossroads. Under these circumstances, the discovery of a new strategy for DDS research would be the responsibility of DDS researchers. Here, we have proposed one possible new strategy for DDS research, the EAT System. No cell can live without energy. Cancer cells are no exception. Additionally, it is a critical to promote “reverse translational research” on the limits of EPR effects observed clinically, and to collect an abundance of valuable clinical data on high-molecular-weight anticancer drugs. Clinical and basic researchers have verified various pathophysiological data, and will continue to pursue the possibility of further DDS research based on clinical data, such as stratification, or individualized treatment of cancer pathology.
Acknowledgments
We thank Dr. Ryo Kinoshita at Kumamoto University for his important contributions to these experiments. This work was supported, in part, by Grants-in-Aid from the Japan Society for the Promotion of Science (JSPS), and a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (KAKENHI KIBAN (B) 18H02587), Japan.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Hollis C. P., Weiss H. L., Leggas M., Evers B. M., Gemeinhart R. A., Li T., J. Contr. Rel., 172, 12–21 (2013).
- 2) Nichols J. W., Bae Y. H., J. Contr. Rel., 190, 451–464 (2014).
- 3) Ishima Y., Sawa T., Kragh-Hansen U., Miyamoto Y., Matsushita S., Akaike T., Otagiri M., J. Pharmacol. Exp. Ther., 320, 969–977 (2007).
- 4) Ishima Y., Akaike T., Kragh-Hansen U., Hiroyama S., Sawa T., Maruyama T., Kai T., Otagiri M., Biochem. Biophys. Res. Commun., 364, 790–795 (2007).
- 5) Ishima Y., Akaike T., Kragh-Hansen U., Hiroyama S., Sawa T., Suenaga A., Maruyama T., Kai T., Otagiri M., J. Biol. Chem., 283, 34966–34975 (2008).
- 6) Ishima Y., Hiroyama S., Kragh-Hansen U., Maruyama T., Sawa T., Akaike T., Kai T., Otagiri M., Nitric Oxide, 23, 121–127 (2010).
- 7) Ishima Y., Shinagawa T., Yoneshige S., Kragh-Hansen U., Ohya Y., Inomata Y., Kai T., Otagiri M., Maruyama T., Nitric Oxide, 30, 36–42 (2013).
- 8) Oshiro S., Ishima Y., Maeda H., Honda N., Bi J., Kinoshita R., Ikeda M., Iwao Y., Imafuku T., Nishida K., Miyamura S., Watanabe H., Otagiri M., Maruyama T., J. Pharm. Sci., 107, 848–855 (2018).
- 9) “Albumin in Medicine: Pathological and Clinical Applications,” ed. by Otagiri M., Chuang V.T.G., Springer, Singapore, 2016.
- 10) Ikuta S., Chuang V. T. G., Ishima Y., Nakajou K., Furukawa M., Watanabe H., Maruyama T., Otagiri M., J. Control. Release, 147, 17–23 (2010).
- 11) Furukawa M., Tanaka R., Chuang V. T. G., Ishima Y., Taguchi K., Watanabe H., Maruyama T., Otagiri M., J. Control. Release, 154, 189–195 (2011).
- 12) Tanaka R., Watanabe H., Kodama A., Chuang V. T. G., Ishima Y., Hamasaki K., Tanaka K., Mizushima T., Otagiri M., Maruyama T., J. Pharmacol. Exp. Ther., 345, 271–283 (2013).
- 13) Tanaka R., Ishima Y., Enoki Y., Kimachi K., Shirai T., Watanabe H., Chuang V. T. G., Maruyama T., Otagiri M., Front. Immunol., 5, 561 (2014).
- 14) Tanaka K. I., Shimoda M., Chuang V. T. G., Nishida K., Kawahara M., Ishida T., Otagiri M., Maruyama T., Ishima Y., Int. J. Pharm., 535, 140–147 (2018).
- 15) Matsumura Y., Maeda H., Cancer Res., 46, 6387–6392 (1986).
- 16) Katayama N., Nakajou K., Ishima Y., Ikuta S., Yokoe J., Yoshida F., Suenaga A., Maruyama T., Kai T., Otagiri M., Nitric Oxide, 22, 259–265 (2010).
- 17) Ishima Y., Hara M., Kragh-Hansen U., Inoue A., Suenaga A., Kai T., Watanabe H., Otagiri M., Maruyama T., J. Control. Release, 164, 1–7 (2012).
- 18) Ishima Y., Kragh-Hansen U., Maruyama T., Otagiri M., Biomed. Res. Int., 2013, 353892 (2013).
- 19) Ishima Y., Fang J., Kragh-Hansen U., Yin H., Liao L., Katayama N., Watanabe H., Kai T., Suenaga A., Maeda H., Otagiri M., Maruyama T., J. Pharm. Sci., 103, 2184–2188 (2014).
- 20) Ishima Y., Inoue A., Fang J., Kinoshita R., Ikeda M., Watanabe H., Maeda H., Otagiri M., Maruyama T., Cancer Sci., 106, 194–200 (2015).
- 21) Ikeda M., Ishima Y., Chuang V. T. G., Ikeda T., Kinoshita R., Watanabe H., Ishida T., Otagiri M., Maruyama T., Nitric Oxide, 69, 28–34 (2017).
- 22) Ishima Y., Biol. Pharm. Bull., 40, 128–134 (2017).
- 23) Ishima Y., Yoshida F., Kragh-Hansen U., Watanabe K., Katayama N., Nakajou K., Akaike T., Kai T., Maruyama T., Otagiri M., Free Radic. Res., 45, 1196–1206 (2011).
- 24) Ishima Y., Kinoshita R., Ikeda M., Kragh-Hansen U., Fang J., Nakamura H., Chuang V. T. G., Tanaka R., Maeda H., Kodama A., Watanabe H., Maeda H., Otagiri M., Maruyama T., J. Control. Release, 217, 1–9 (2015).
- 25) Ishima Y., Chen D., Fang J., Maeda H., Minomo A., Kragh-Hansen U., Kai T., Maruyama T., Otagiri M., Bioconjug. Chem., 23, 264–271 (2012).
- 26) Ishima Y., Kinoshita R., Chuang V. T. G., Nakamura H., Fang J., Watanabe H., Shimizu T., Okuhira K., Ishida T., Maeda H., Otagiri M., Maruyama T., Biomaterials, 140, 162–169 (2017).
- 27) Yoshikawa T., Mori Y., Feng H., Phan K. Q., Kishimura A., Kang J. H., Mori T., Katayama Y., Int. J. Pharm., 565, 481–487 (2019).
- 28) Islam W., Fang J., Imamura T., Etrych T., Subr V., Ulbrich K., Maeda H., Mol. Cancer Ther., 12, 2643–2653 (2018).
- 29) Schnitzer J. E., Oh P., J. Biol. Chem., 269, 6072–6082 (1994).
- 30) Tiruppathi C., Finnegan A., Malik A. B., Proc. Natl. Acad. Sci. U.S.A., 93, 250–254 (1996).
- 31) Iancu C., Mocan L., Bele C., Orza A. I., Tabaran F. A., Catoi C., Stiufiuc R., Stir A., Matea C., Iancu D., Agoston-Coldea L., Zaharie F., Mocan T., Int. J. Nanomedicine, 6, 129–141 (2011).
- 32) Schnitzer J. E., Allard J., Oh P., Am. J. Physiol., 268, H48–H55 (1995).
- 33) Tiruppathi C., Song W., Bergenfeldt M., Sass P., Malik A. B., J. Biol. Chem., 272, 25968–25975 (1997).
- 34) Sage H., Vernon R. B., Funk S. E., Everitt E. A., Angello J., J. Cell Biol., 109, 341–356 (1989).
- 35) Jacob K., Webber M., Benayahu D., Kleinman H. K., Cancer Res., 59, 4453–4457 (1999).
- 36) Lane T. F., Sage E. H., FASEB J., 8, 163–173 (1994).
- 37) Pichler R. H., Bassuk J. A., Hugo C., Reed M. J., Eng E., Gordon K. L., Pippin J., Alpers C. E., Couser W. G., Sage E. H., Johnson R. J., Am. J. Pathol., 148, 1153–1167 (1996).
- 38) Desai N., Trieu V., Damascelli B., Soon-Shiong P., Transl. Oncol., 2, 59–64 (2009).
- 39) Kratz F., J. Control. Release, 132, 171–183 (2008).
- 40) Kamphorst J. J., Nofal M., Commisso C., Hackett S. R., Lu W., Grabocka E., Vander Heiden M. G., Miller G., Drebin J. A., Bar-Sagi D., Thompson C. B., Rabinowitz J. D., Cancer Res., 75, 544–553 (2015).
- 41) Seno T., Harada H., Kohno S., Teraoka M., Inoue A., Ohnishi T., Int. J. Oncol., 34, 707–715 (2009).
- 42) Larsen M. T., Kuhlmann M., Hvam M. L., Howard K. A., Mol. Cell Ther., 4, 3 (2016).
- 43) Sleep D., Expert Opin. Drug Deliv., 12, 793–812 (2015).
- 44) Chatterjee M., Ben-Josef E., Robb R., Vedaie M., Seum S., Thirumoorthy K., Palanichamy K., Harbrecht M., Chakravarti A., Williams T. M., Cancer Res., 77, 5925–5937 (2017).