Introduction
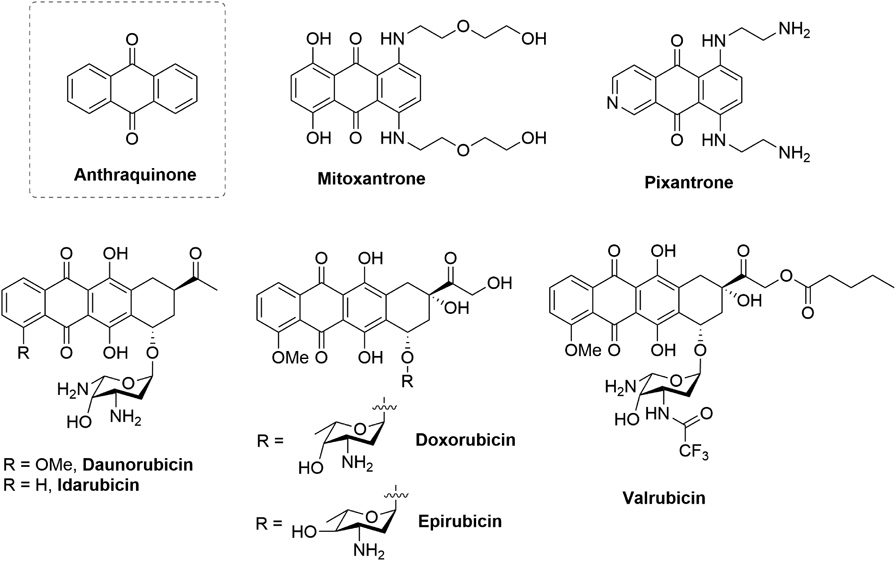
Anthraquinones are an important class of both natural and synthetic compounds with a broad bioactivity, including anticancer,1) antibacterial,2) neuroprotective effects3) and so on.4) The scaffold of anthraquinones is a tricyclic diketone structure as one of important pharmacophores for treating cancers4) (Fig. 1). For examples, mitoxantrone5) and pixantrone6) are synthetic analogs of anthraquinone, which were approved anticancer drugs by the Food and Drug Administration (FDA) in U.S.A., or Europe, respectively. Anthracycline antibiotics with the pharmacophore of anthraquinone are an important class of anticancer drugs with high potency against various solid and hematological malignancies.4) Since the first discovery of daunorubicin produced as a secondary metabolite by Streptomyces peucetius with a remarkable anticancer activity,7,8) many of natural, semisynthetic and synthetic analogs have been extensively investigated for their biological and clinical properties.4,9) So far five of them (daunorubicin, doxorubicin, epirubicin, idarubicin, and valrubicin) have been approved by FDA for the treatment of cancers.4,9) Despite their outstanding anticancer efficacy, there is increasing concern and recognition of some side effects like mutagenicity, genotoxicity and cardiotoxicity.10,11) Although the exact mechanism of these side effects is still not clear, disrupting DNA and RNA synthesis by noncovalent binding to DNA and thus inhibiting topoisomerase II of DNA repair mechanisms are believed to contribute mainly inducing these side effects. Therefore, more anthraquinone derivatives with better efficacy but less safety issues are definitely to be expected.
On the other hand, histone deacetylase (HDAC) inhibitors are a class of targeted pharmaceuticals for treating cutaneous or peripheral T-cell lymphoma, or multiple myeloma with less side effects excepting of QT interval prolongation.11,12) Up to date, five HDAC inhibitors (Fig. 2) have already been approved to the market for the treatment of human cancers including Vorinostat (SAHA),13) Romidepsin (FK228)14) and Belinostat (PXD-101)15) and Panobinostat (LBH-589)16) by FDA, Chidamide17) by China Food and Drug Administration (CFDA). The structural feature of HDAC inhibitors consists of zinc binding group (ZBG), linker and Cap group.18) Usually, a cap group is the bulky group. Although HDAC is a good therapeutic target for achieving anticancer efficacy with less safety issues, few anthraquinone derivates focus on this target.19,20)
Since topoisomerase II and HDAC are colocalized in the nucleus, the suitable modification of anthraquinone derivates will facilitate their targeting HDAC.21–23) In view of the mechanism of HDAC inhibitors, ZBG plays a decisive role in HDAC inhibition, which can form the tight binding with zinc ion in the catalytic pocket of HDAC. We expected that this kind of tight binding if anthraquinone derivates as HDAC inhibitors may prevent them interacting with topoisomerase II and thus switch therapeutic target from topoisomerase II to HDAC, to avoid their side effects in cancer treatment like cytotoxicity and cardiotoxicity. Once the tight binding is forming, their effect on TOP II may decrease logically and theoretically. Considering on this, we would like to take advantage of anthraquinone as a cap group by assembling ZBG to design a serial of HDAC inhibitors to minimize those inherent side effects.
Results and Discussion
To test this idea, we firstly used 1-amino anthraquinone 1 as a starting material, which was coupled with a series of different dicarboxylic acids and then converted into hydroxamates 4. Similarly, hydroxamates 7 were assembled by the coupling and conversion of 2-amino anthraquinone 5 and a series of different dicarboxylic acids. Considering the length of the linker of SAHA, we fixed the length of the linker by the coupling of 2-amino anthraquinone 1 and 1, 8-octanedioic acid and then assembled different groups to give candidate compounds 9 for investigating any potential ZBG groups. We also take the reference of the structure of Chidamide to couple successively 1-amino anthraquinone 1 with glutaric acid, p-carboxybenzylamine and 4-fluorobenzene-1,2-diamine (or benzene-1,2-diamine or hydroxylamine) to obtain candidate compounds 12. The details for the synthesis and identification of candidates (4, 7, 9, and 12) are shown in the Supplementary Materials.
With these candidates in hand, we then did the pilot screening for general HDAC inhibition at a compound’s concentration of 1 µM. All tested compounds were subjected to the deacetylation assay by using a nuclear extract of HeLa cells as a source of HDACs and Boc-Ac-Lys-7-amino-4-methylcoumarin (AMC) as a fluorogenic substrate. As a result, the HDAC inhibition was increasing along with the length of the linker in compounds 4 goes up from compound 4a to 4d (the inhibition from 72.6 to 101.5%, Table 1). The HDAC inhibition was decreased to 92.4% by increasing the length of the linker in compound 4e while the inhibition was abolished when the further increasing the length of the linker in compound 4f (7.5%, Table 1). These results indicated that the favorable linkers of compounds 4c–e was those derivatized from adipic acid, pimelic acid and suberic acid, respectively (Fig. 3). Similarly, the inhibition was increasing from 45.5 to 101.7% when the length of the linker in compounds 7 goes up from that of compound 7a to 7d (Table 1). The replacing the hydroxamate group of compound 4d to other potential ZBG groups showed no obvious effects on the HDAC inhibition (9a–k vs. 4d, Table 1). For the mimics of Chidamide 12a–c, only 12a show a slight inhibition while 12c show a moderate inhibition (Table 1). To further evaluate the inhibitory potency, we then measured their IC50 values. As shown in Table 1, the IC50 values of compounds 4 and 7 varied from 8.6 ± 1.3 to 310.0 ± 1.2 nM. Among them, several compounds (4c, d and 7b–d) show better inhibition than that of SAHA, falling into the nanomolar range with the IC50 values from 8.6 ± 1.3 to 34.7 ± 1.5 nM (4c, d and 7b–d vs. SAHA, Table 1). Compound 4d was eventually turned out to be the most potent HDAC inhibitor with the IC50 value of 8.6 ± 1.3 nM.
Table 1. HDAC Inhibition for Candidates (
4,
7,
9, and
12)
| Cmpd | Inhibition at 1 µM/% (IC50/nM) | Cmpd | Inhibition at 1 µM/% (IC50/nM) |
|---|
| 4a | 72.6 (310.0 ± 1.2) | 9d | —* |
| 4b | 72.1 (205.5 ± 0.9) | 9e | —* |
| 4c | 98.5 (26.1 ± 2.4) | 9f | —* |
| 4d | 101.5 (8.6 ± 1.3) | 9g | —* |
| 4e | 92.4 (71.2 ± 0.6) | 9h | —* |
| 4f | 7.5 | 9i | 25.0 |
| 7a | 45.5 | 9j | —* |
| 7b | 97.2 (34.7 ± 1.5) | 9k | —* |
| 7c | 97.7 (25.9 ± 2.1) | 12a | 38.1 |
| 7d | 101.7 (12.1 ± 0.8) | 12b | —* |
| 9a | 11.8 | 12c | 48.8 |
| 9b | —* | SAHA | 92.2 (50.0 ± 0.4) |
| 9c | 6.3 | | |
To further evaluate the inhibitory potency and selectivity of some compounds, we measured their IC50 values using the activity assay kits of HDAC1, 2 and 3 as Class I HDACs and HDAC6 as a Class II HDAC (Table 2). All tested compounds 4b, 4d, 7b, and 7d demonstrated the comparable inhibitory potency against HDAC6 with IC50 values from 4.66 to 12.46 nM while those IC50 values in two-digital of nano-molar range against HDAC1/2/3. Overall, these compounds seem to be non-selective HDAC inhibitors as a classic pan-HDAC inhibitor, SAHA. Before the cellular study, we also evaluated the drug-like features of compounds 4b, 4d, 7b, and 7d in comparison of that of SAHA, which was performed by means of FAFDrugs.20,24) Like SAHA, the data of compounds 4b, 4d, 7b, and 7d showed satisfactory results, displaying suitable predicted cLogP and well obeying Lipinski’s rule of five and Veber’s criteria, suggesting that it could be orally absorbed, however further experiments were still needed to confirm these properties (Table S1 in Supplementary Materials).
Table 2. IC
50 Values (nM) of Compounds
4b,
4d,
7b, and
7d against Class I and Class II HDACs
| IC50 Values/nM |
|---|
| Cmpd | HDAC1 | HDAC2 | HDAC3 | HDAC6 |
|---|
| 4b | 70.1 ± 1.3 | / | / | 11.5 ± 0.6 |
| 4d | 7.7 ± 2.1 | 41.4 ± 0.4 | 12.6 ± 1.3 | 5.9 ± 0.2 |
| 7b | 29.7 ± 0.4 | 104.2 ± 3.4 | 41.6 ± 1.0 | 11.4 ± 1.0 |
| 7d | 51.1 ± 0.2 | 96.4 ± 0.6 | 39.0 ± 0.2 | 5.1 ± 0.4 |
| SAHA | 93.6 ± 1.7 | >200 | 89.7 ± 0.6 | 20.2 ± 0.9 |
Next, we further evaluate whether compounds 4b, 4d, 7b, and 7d could work on the cellular HDACs (Fig. 4). Therefore, these compounds were engaged in Western blotting analysis to evaluate the acetylation levels of α-tubulin. The human breast cancer MCF-7 and leukemia K562 cell lines were treated with control (dimethyl sulfoxide (DMSO)) or compounds 4b, 4d, 7b, and 7d at their indicated concentrations for 8 h, respectively, in comparison with SAHA at 10 µM as a positive control. As shown in Fig. 3, these compounds all induced a dose-dependent increase in acetylation levels of α-tubulin in both MCF-7 and K562 cell lines, suggesting that they could inhibit HDAC in cells. Notably, the cellular inhibition of compounds 4b and 4d derived from 1-amino anthraquinone 1 was superior to that of compound 7d derived from 2-amino anthraquinone 5 (Fig. 4).
Satisfied with their in vitro and cellular inhibition, we then investigated the effect of compounds 4b, 4d, 7b, and 7d on the proliferation of K562, MCF-7 and HeLa cancer cells, which were treated with the indicated compounds at various concentrations for 2 d and the IC50 values were calculated from the measured cell viability using CCK-8 assay as previously described.25) As shown in Table 3, all tested compounds demonstrated comparable antiproliferative activities with the IC50 values in several micro-molar scales in three cancer cell lines, which were close to the effect of SAHA. Although mitoxantrone demonstrated the impressive efficacy of the antiproliferation on these cancer cell lines, mitoxantrone like other anthraquinone derivatives also showed the equally distinguished antiproliferative activity on H9c2 cell lines (one non-cancer cell line, cardiomyocyte), resulting in some side effects of many anthraquinone derivatives like cardiotoxicities.10,11) Interestingly, compound 4b displayed certain selectivity (approx. 3 fold) of antiproliferative effects on cancer cell lines over non-cancer cell lines (Table 3), indicating that anthraquinone derivatives targeting HDAC may reduce their inherent side effects.
Table 3. The Antiproliferative IC
50 Values of Compounds
4b,
4d,
7b, and
7d against K562, MCF-7, HeLa Cells and H9c2 Using CCK-8 Assay
| IC50 Values/µM |
|---|
| Cmpd | K562 | MCF-7 | HeLa | H9c2 |
|---|
| 4b | 9.7 ± 1.1 | 10.6 ± 1.8 | 12.0 ± 1.0 | 31.2 ± 1.5 |
| 4d | 5.3 ± 1.2 | 5.3 ± 0.2 | 7.5 ± 1.7 | 10.0 ± 1.7 |
| 7b | 6.0 ± 2.6 | 10.4 ± 0.4 | 5.0 ± 1.1 | 5.0 ± 0.5 |
| 7d | 2.5 ± 1.3 | 3.4 ± 0.4 | 3.0 ± 1.3 | 2.0 ± 0.2 |
| SAHA | 1.9 ± 0.2 | 2.6 ± 0.6 | 2.7 ± 0.2 | 4.6 ± 0.4 |
| Mitoxantrone | 0.026 | 0.35 | 0.082 | 0.054 |
Finally, we detected their effects on inducing apoptosis and cell cycle by flow cytometry in K562 and MCF-7 cells. Based on the IC50 values of compounds 4b, 4d and 7d, 1, 5 and 10 µM were used as working concentrations for analysis. K562 and MCF-7 cells were treated with control (DMSO) or compounds 4b, 4d and 7d for 24 h and then the cells were stained with Annexin V+/propidium iodide (PI)+ or PI by using flow cytometer to analyze the percentage of apoptosis cells and cell cycle phase distribution. The results showed that, all tested compounds can significantly induce the apoptosis in a dose-dependent manner in these two cancer cell lines (Figs. 5A, 5B). In terms of cell cycle, these compounds’ behavior seems quite distinction in different cells. For an example, after exposure to 4b, the G2-arrested was observed in K562 cells while the S1-arrested was found in MCF-7 cells (Figs. 5C, 5D). In addition, they all demonstrated the cell cycle phase arrested in a concentration-dependent way.
Since anthraquinones are attractive scaffolds to develop useful compounds for treating cancers, anthraquinone derivatives have been extensively investigated. However, most of them have inherent side effects, mainly due to their inhibition of topoisomerase II of DNA repair mechanisms. Therefore, anthraquinone derivatives targeting other therapeutic targets are still to be expected. We herein described a series of anthraquinone derivatives designed to preferentially inhibit HDACs. This goal was reached by using a tricyclic diketone of anthraquinone as a cap group. Our focused design strategy ultimately led to compounds 4b, 4d, 7b, and 7d displaying the comparable inhibition in enzymatic activity and cell proliferation than that of SAHA, especially, compound 4b displayed certain selectivity (approx. 3 fold) of antiproliferative effects on cancer cell lines over non-cancer cell lines (Table 3), suggesting that anthraquinone derivatives targeting HDAC may reduce their inherent side effects.10,11) The strategy by switching therapeutic target from topoisomerase II to HDAC in the design of anthraquinone derivatives has shown promising results in treating cancer cells. Further studies and compound optimization are undergoing in our laboratories.