Regular Articles
Dioscin Protects against Aβ1–42 Oligomers-Induced Neurotoxicity via the Function of SIRT3 and Autophagy
2020 年 68 巻 8 号 p. 717-725
詳細
2020 年 68 巻 8 号 p. 717-725
Alzheimer’s disease (AD) is a common neurodegenerative disease with high incidence among old people. Dioscin is a product extracted from natural herbs, which has multiple pharmacological activities. In this study, we investigated the potential effects of disocin on amyloid-β peptide (Aβ1–42) oligomers-treated HT22 cells. Aβ1–42 oligomers induced great neurotoxicity to HT22 cells as examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The results of terminal deoxynucleoitidyl transferase-mediated deoxyuridine triphosphate biotin nich end labeling (TUNEL) staining and flow cytometry indicated that Aβ1–42 oligomers led to increased apoptosis and generation of reactive oxygen species (ROS) in HT22 cells. However, dioscin could remarkably inhibit the neurotoxicity induced by Aβ1–42 oligomers, as well as decrease the apoptosis and ROS generation. Sirtuin-3 (SIRT3) staining and quantification indicated that dioscin upregulated the expression of neuroprotective SIRT3. Moreover, dioscin induced the formation of autophagosomes and autolysosomes in HT22 cells. Dioscin also enhanced the levels of Beclin-1 and LC3-II while decreased the level of p62. These results suggested that dioscin could activate autophagy in HT22 cells. It was also found that knocking down SIRT3 resulted in the downregulation of Beclin-1, LC3-II and the aggregation of p62, suggesting that SIRT3 was an important regulator in autophagy. Furthermore, we found that knocking down SIRT3 or inhibiting autophagy suppressed the protective effects of dioscin on Aβ1–42 oligomers-induced neurotoxicity, apoptosis and ROS generation. These results revealed that SIRT3 and autophagy functioned together in the neuroprotective mechanisms of dioscin. Therefore, dioscin might be a promising drug to protect against Aβ1–42 oligomers-induced neurotoxicity and reduce neuron damage or death in AD.
Alzheimer’s disease (AD) is a progressive neurodegenerative disease which blames to the major occurrence of dementia. AD develops with the increase of age and has a much higher incidence among people aged over 70 years old.1) Those affected with AD usually have difficulties in cognition, communication and action, especially when the disease progresses to a severe stage.2) Two pathological changes in the cerebra of Alzheimer’s patients have been identified as the hallmarks of AD, including the formation of sufficient senile plaques and neurofibrillary tangles, which result from the accumulation of extracellular amyloid-β peptide (Aβ) and the aggregation of intracellular twisted protein tau.3,4) The extracellular Aβ plaques contribute to synaptic dysfunction by interfering with the communication from neuron to neuron, while the intracellular tau tangles damage the stability of microtubule and block the transport of nutrients inside the neurons.1) Drugs of rivastigmine, galantamine and donepezil are acetylcholinesterase inhibitors that can slow down the decomposition of acetylcholine and enhance the memory of Alzheimer’s patients.5) However, the effectiveness of these drugs is temporary and no drug has been proved to stop or reverse the process of AD at present.
Autophagy is a degradative pathway to eliminate abnormal proteins or damaged organelles within the cells, which plays an important role in maintaining the intracellular homeostasis.6) Three general subtypes of autophagy have been identified in most mammalian cells so far, including macroautophage, microautophage and chaperone-mediated autophagy. Macroautophage is a dominant catabolic mechanism for eukaryotic cells to degrade the intracellular proteins and organelles.7) In the process of macroautophage, cytoplasmic constituents are isolated in a double-membrane autophagosome, delivered to the lysosome and then digested by a range of acidic hydrolases.8) Nevertheless, emerging evidences implicate the dysfunction of autophagy in some neurodegenerative diseases, such as Parkinson’s disease and AD, which may contribute to the pathogenesis of these diseases.8,9) Thus, activation of autophagy is regarded as a promising approach to treat AD, through which the aggregated Aβ and tangled protein tau may be selectively eliminated. Sirtuin-3 (SIRT3) is a deacetylase that involves in many aspects of mitochondrial biology, including reactive oxidative pathways and energy metabolism processes.10,11) Recent researches show that SIRT3 may have neuroprotective effects on AD and promote autophagy by regulating several autophagy-related genes in neuronal ischemia.12,13) But it has not been clarified that whether SIRT3 can protects against AD through activating the process of autophagy.
Dioscin is a steroidal saponin originated from some natural herbs, which has multiple biological activities of anti-inflammation, anti-tumor and anti-allergy.14,15) Dioscin was found to alleviate fructose-induced renal damage through activating SIRT3-mediated signaling pathway.16) Dioscin also had neuroprotective effects in a rat model of cerebral ischemia/reperfusion.17) In addition, dioscin could induce the autophagy in human lung cancer cells.18) However, the effect of dioscin on AD has not been reported yet. It is unclear that whether dioscin could protect against AD via the regulation of SIRT3. In this study, we established in vitro HT22 hippocampal neuronal cell model to investigate the possible effects of dioscin on treating AD and explore the underlying mechanisms.
Aβ1–42 oligomers were synthesized according to previously reported method.19) Briefly, 1 mg Aβ1–42 (Glbiochem, China) was dissolved in 220 µL hexafluoroisopropanol. Then the solution was removed to the fume hood for volatilization of hexafluoroisopropanol. After a peptide film was formed, 44 µL 100% dimethyl sulfoxide (DMSO) was added to dissolve it and Aβ1–42 solution with the concentration of 5 mmol/L was finally obtained. The Aβ1–42 solution was diluted to 40 µmol/L and incubated at 4°C for 24 h. After centrifuging at 1400 rpm for 10 min, the supernatant was obtained as Aβ1–42 oligomers. The Aβ1–42 oligomers were charaterized by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue (Solarbio, China) staining.
Cell Culture and Optimization of Dioscin ConcentrationHT22 cells (Zqxzbio, China) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, U.S.A.) added with 10% fetal bovine serum (Hyclone, U.S.A.) and incubated at 37°C in an atmosphere of 95% air/5% CO2. The cells were used when they grew into the density of 80–90%. To optimize the concentration of dioscin (MedChemExpress, HY-N0124, China), cells were pretreated with 100, 200, 400, 800 or 1600 nmol/L dioscin for 1 h and then added with 1 µmol/L Aβ1–42 oligomers to incubate for 48 h. Cells in Aβ1–42 group were only treated with 1 µmol/L Aβ1–42 oligomers for 48 h. The optimal concentration of dioscin were determined by cell proliferative activity. The further experiments were conducted basing on the optimal concentration of dioscin.
Cell Transfection and Drug TreatmentHT22 cells were treated with the mixture of 200 µL Opti-MEM, 6 µL RNAiMAX (Invitrogen, U.S.A.) and 2 µg SIRT3 small interfering RNA (siRNA) (or NC siRNA as negative control) for transfection. After incubating for 6 h, the culture medium was replaced and 400 nmol/L dioscin or 3-methyladenine (3-MA, MedChemExpress, China) solution was added to incubate for 1 h. Then the medium was supplemented with 1 µmol/L Aβ1–42 oligomers to culture for another 48 h.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) AssayMTT assay was performed to assess the proliferative activity of HT22 cells after exposing to different drug treatments. HT22 cells were grouped and seeded into 96-well plates. After exposing to corresponding drug treatments, the cells were incubated in complete DMEM medium containing 0.5 mg/mL MTT (KeyGen Biotech, China) for 5 h. Then 150 µL DMSO solution was added to dissolve the formazan generated in the cells. Ten minutes later, the optical density (OD) values were determined by a Microplate Reader (BIOTEK, ELX-800, U.S.A.) at 570 nm.
Terminal Deoxynucleoitidyl Transferase-Mediated Deoxyuridine Triphosphate Biotin Nich End Labeling (TUNEL)HT22 cells were treated with 0.1% Triton X-100 (Beyotime, China) at room temperature for 15 min to permeabilize the cells and then washed by phosphate buffer saline (PBS) for 3 times. For TUNEL staining, cells were incubated with TUNEL reagents at 37°C for 60 min according to instructions of the assay kit (In Situ Cell Detection Kit, Roche, Switzerland) and washed by PBS for 3 times. 4′,6-Diamidino-2-phenylindole (DAPI) (Beyotime) was then used to stain the nuclei of HT22 cells for 5 min. After washing with PBS for 3 times, images were captured under a fluorescence microscope (OLUMPUS, BX53, Japan).
Flow CytometryHT22 cells were seeded into 6-well plates and conducted with corresponding transfection and drug treatments. The cells were collected and suspended in the binding buffer with a density of 1 × 106 cells/mL. The apoptotic HT22 cells were detected with an assay kit (Apoptosis Detection Kit, Solarbio) and analyzed by a flow cytometer (ACEA Biosciences, NovoCyte, U.S.A.).
Quantification of Reactive Oxygen Species (ROS)HT22 cells were collected and centrifuged for 5 min at 140 × g. After removing the supernatant, the cells were mixed with 1 mL 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) solutions (1 : 1000 diluted by serum-free medium) and incubated at 37°C for 20 min according to instructions of the assay kit (Beyotime). The cells were washed by PBS for 3 times, resuspended in PBS and determined by the flow cytometer.
Real-Time Quantitative PCRReal-time quantitative PCR was used to analyze the relative mRNA level of SIRT3. Briefly, total RNA was extracted from HT22 cells using the RNA isolation kit (Tiangen, China) according to the manufacture’s instruction. The RNA concentration of different samples was determined by an Ultraviolet Spectrophotometer (Thermo Scientific, NANO 2000, U.S.A.). Then reverse transcription was performed by a real-time PCR instrument (BIONEER, Exicycler 96, Korea) to synthesize the cDNA template. Quantitative real-time PCR reaction was then performed on the PCR system according to the instruction. The forward and reverse primer sequences used in PCR were listed as follows: SIRT3 forward 3′-CCC CGA CTG CTC ATC AA-5′, SIRT3 reverse 3′-CCA CCA GCC TTT CCA CA-5′, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward 3′-TGT TCC TAC CCC CAA TGT GTC CGT C-5′, GAPDH reverse 3′-CTG GTC CTC AGT GTA GCC CAA GAT G-5′.
Immunofluorescence Staining of SIRT3HT22 cells were fixed with 4% paraformaldehyde for 15 min and permeabilized in 0.1% Triton X-100 for 30 min. The cells were washed by PBS for 3 times and then immersed in goat serum (Solarbio) for 15 min. Immunofluorescence staining was performed by incubating HT22 cells with SIRT3 antibody (Abclonal, China) at 4°C overnight and Cy3-labeled Goat Anti-Rabbit immunoglobulin G (lgG) (Beyotime) at room temperature for 60 min. The primary and secondary antibodies were diluted by PBS at a ratio of 1 : 200 before use. DAPI was then used to stain the nuclei of these cells. The images were captured under the fluorescence microscope.
Infection with Monomeric Cherry-Green Fluorescent Protein-LC3 (mCherry-GFP-LC3) Adenoviral VectorsHT22 cells were cultured in 24-well plates and infected with mCherry-GFP-LC3 adenoviral vectors (provided by HanBio Technology, China) according to the protocols of the manufacturer. After incubating for 24 h, the HT22 cells were performed with corresponding treatments of dioscin and Aβ1–42 oligomers and observed under the fluorescence microscope. The green fluorescence of GFP was diminished in acidic condition while the red fluorescence of mCherry remained stable. The red puncta and yellow puncta were respectively identified as autolysosomes and autophagosomes in the cells.
Western BlotHT22 cells were collected and lysed in radio immunoprecipitation assay (RIPA) lysis buffer (Solarbio) containing 1 mM phenylmethanesulfonyl fluoride (Solarbio) to extract the total protein. The protein concentration was determined by a bicinchoninic acid (BCA) protein assay kit (Solarbio). Equal amount of protein in different groups was separated on polyacrylamide gels under an electrophoretic voltage of 80 V for 2.5 h, and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, U.S.A.). The PVDF membrane was blocked by 5% non-fat milk solution for 1 h and incubated with primary antibodies at 4°C overnight and horseradish peroxidase (HRP)-coupled secondary antibodies at 37°C for 1 h. After washing for 3 times, the membrane was treated with ECL chemiluminescent reagent (Solarbio) for 5 min and exposed in darkroom to detect the protein bands. The optical intensity was calculated by the software (Gel-Pro-Analyzer) and normalized to the control of GAPDH. The primary and secondary antibodies were listed in Table 1.
| Antibody | Diluted ratio | Source |
|---|---|---|
| SIRT3 | 1 : 1000 | Proteintech, China |
| Beclin-1 | 1 : 5000 | |
| p62 | 1 : 2000 | |
| GAPDH | 1 : 10000 | |
| LC3-I/II | 1 : 1000 | Affinity, China |
| Goat anti-rabbit IgG-HRP | 1 : 3000 | Solarbio, China |
| Goat anti-mouse IgG-HRP | 1 : 3000 |
All data were represented as mean ± standard deviation (n = 3). The significance between two or more groups were analyzed by GraphPad Prism software. Two-tailed Student’s t-test was applied to calculate the significance between two groups. One-way ANOVA was applied to calculate the significance between three or more groups and Tukey’s post-hoc test was used for multiple comparisons.
The composition of synthesized Aβ1–42 oligomers was characterized by SDS-PAGE. Two main protein bands with the molecular weight around 12 and 16 kDa were identified in Aβ1–42 oligomers, showing the composition of trimers and tetramers (Fig. 1A). To simulate the neurol death in AD, Aβ1–42 oligomers was used to induce the neurotoxicity in HT22 cells. As examined by MTT assay, the proliferative activity was significantly decreased in HT22 cells of Aβ1–42 group compared with normal Control group (Fig. 1B), confirming that Aβ1–42 oligomers induced great neurotoxicity in HT22 cells. In order to compare the effect of dioscin with different concentration, we pretreated HT22 cells with 100, 200, 400, 800 or 1600 nmol/L dioscin before they exposed to Aβ1–42 oligomers. The result of MTT assay showed that 400, 800 and 1600 nmol/L dioscin had obvious effects on reducing Aβ1–42 oligomers-induced neurotoxicity and enhancing proliferative activity of HT22 cells while 100 nmol/L and 200 nmol/L dioscin had no significant effect, comparing to Aβ1–42 group (Fig. 1B). Moreover, there was no significant difference among the effect of 400, 800 and 1600 nmol/L dioscin. Therefore, 400 nmol/L dioscin was chosen for the further study. We next confirmed that 400 nmol/L dioscin had a sufficient effect at 48 h since the proliferative activity could not be greatly enhanced at 72 h (Fig. 1C). The further experiments were conducted based on the optimized concentration of dioscin and reaction time. Representative images of TUNEL staining for apoptosis exhibited that the apoptotic cells in Aβ1–42 + dioscin group seemed to be less than in Aβ1–42 group (Fig. 1D). Further, analysis of flow cytometry validated that the apoptotic rate in Aβ1–42 + dioscin group was significantly reduced compared with Aβ1–42 group (Figs. 1E, F). These results demonstrated that dioscin could inhibit Aβ1–42 oligomers-induced apoptosis of HT22 cells. Quantification of ROS by flow cytometry showed that Aβ1–42 oligomers significantly induced the generation of ROS in Aβ1–42 group, while dioscin effectively inhibited the generation of ROS in HT22 cells exposed to Aβ1–42 oligomers (Figs. 1G, H). These results suggested that dioscin could protect against Aβ1–42 oligomers-induced neurotoxicity and reduce the neuron damages.
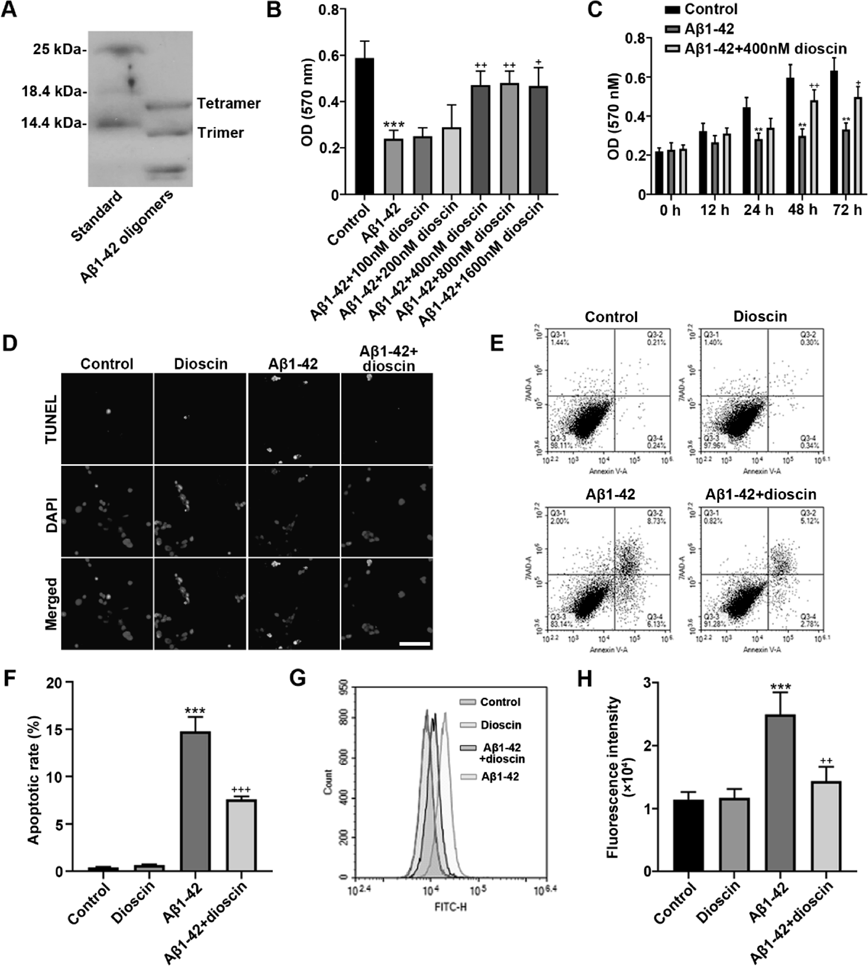
(A) The representative gel of synthetic Aβ1–42 oligomers characterized by SDS-PAGE. (B) Optical density (OD) values determined after MTT assay in HT22 cells treated with Aβ1–42 oligomers and different concentration of dioscin. (C) OD values determined after MTT assay in HT22 cells incubated for different time. (D) Representative images of TUNEL staining. Scale bar = 100 µm. (E) Apoptosis of HT22 cells examined by flow cytometry. (F) Apoptotic rate of HT22 cells. (G) Reactive oxygen species (ROS) generation determined by flow cytometry. (H) Fluorescence intensity of ROS in HT22 cells determined by flow cytometry. * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with Control group. + p < 0.05, ++ p < 0.01 and +++ p < 0.001, compared with Aβ1–42 group.
In previous research, SIRT3 had been proved to have protective effect on neuronal ischemia13) and dioscin could regulate SIRT3-mediated signaling pathway in renal damage.16) But the relationship between dioscin and SIRT3 in neuron cells was not clear. Thus, we performed immunofluorescence staining to detect SIRT3 expression in HT22 cells. Representative images showed that more SIRT3 (red fluorescence) was expressed in Aβ1–42 + dioscin group than in Aβ1–42 group (Fig. 2A), indicating that Aβ1–42 oligomers inhibited the expression of SIRT3 while dioscin could dramatically enhance its level. We also quantified the mRNA and protein expression of SIRT3 by quantitative real-time PCR and Western blot. Results showed that dioscin could significantly upregulate the SIRT3 expression in Dioscin group compared with Control group (Figs. 2B, C). On the contrary, Aβ1–42 oligomers strongly inhibited SIRT3 expression in Aβ1–42 group. Therefore, we confirmed that dioscin could also regulate SIRT3 expression in HT22 neuron cells. More importantly, it was found that SIRT3 expression was remarkably upregulated in Aβ1–42 + dioscin group, demonstrating that dioscin could still enhance the level of SIRT3 in HT22 cells exposed to Aβ1–42 oligomers and the upregulated SIRT3 might contribute to protect against Aβ1–42 oligomers-induced neurotoxicity.
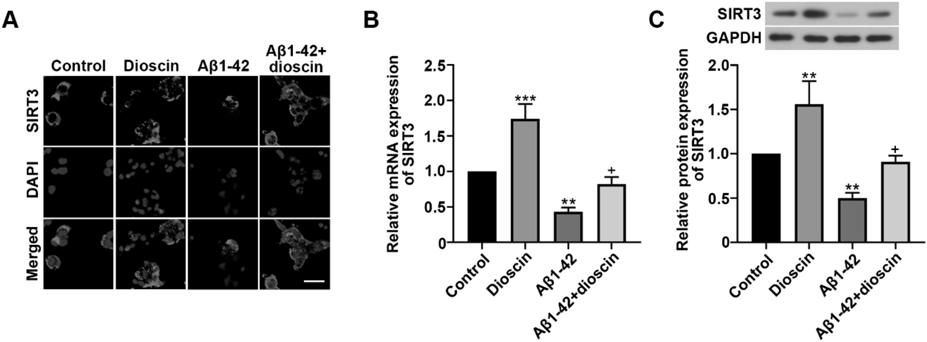
(A) Representative images of immunofluorescence staining for SIRT3. Scale bar = 50 µm. (B) The relative mRNA expression of SIRT3 determined by real-time PCR. (C) The relative protein expression of SIRT3 determined by Western blot. * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with Control group. + p < 0.05, ++ p < 0.01 and +++ p < 0.001, compared with Aβ1–42 group.
HT22 cells transfected with mCherry-GFP-LC3 adenoviral vectors showed much more red puncta (autolysosome) and yellow puncta (autophagosome) in Dioscin group than in Control group (Fig. 3A). On the contrary, less red and yellow puncta could be observed in Aβ1–42 group, indicating that Aβ1–42 oligomers inhibited the formation of autolysosomes and autophagosomes. However, HT22 cells in Aβ1–42 + dioscin group exhibited more puncta of autolysosomes and autophagosomes, demonstrating that dioscin promoted the process of autophagy. We next investigated the autophagy in HT22 cells by analyzing the expression of autophagy-related genes. Beclin-1 and LC3-II have been found to play positive roles in promoting autophagy while inhibition of autophagy leads to the aggregation of p62. Western blot showed that the expression of Beclin-1 and LC3-II was dramatically downregulated in Aβ1–42 group accompanied with the upregulation of p62 (Figs. 3B, C, D, F), implying that Aβ1–42 oligomers inhibited the autophagy in HT22 cells. However, the levels of Beclin-1 and LC3-II were greatly enhanced in Aβ1–42 + dioscin group compared with Aβ1–42 group while the level of p62 was decreased, which suggested that dioscin could promote autophagy in HT22 cells exposed to Aβ1–42 oligomers. The expression of LC3-I had no significant difference among the four groups (Fig. 3E). These results indicated that dioscin could induce autophagy in HT22 cells exposed to Aβ1–42 oligomers and dioscin-induced autophagy might also contribute to protect against Aβ1–42 oligomers-induced neurotoxicity.

(A) Representative fluorescent images showing the location of mCherry-GFP-LC3 in HT22 cells. Scale bar = 25 µm. (B) The protein bands of Beclin-1, LC3-I, LC3-II and p62 determined by Western blot. The relative protein expression of (C) Beclin-1, (D) LC3-II, (E) LC3-I and (F) p62 in HT22 cells. * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with Control group. + p < 0.05, ++ p < 0.01 and +++ p < 0.001, compared with Aβ1–42 group.
To explore the association between autophagy and SIRT3, HT22 cells were transfected with SIRT3 siRNA to knock down SIRT3 expression or transfected with NC siRNA as negative control. It was found that HT22 cells transfected with SIRT3 siRNA expressed much lower SIRT3 compared with NC siRNA group (Figs. 4A, B, C), demonstrating the effectiveness of SIRT3 siRNA. Then HT22 cells transfected with SIRT3 siRNA or NC siRNA were treated with dioscin and Aβ1–42 oligomers. We determined the expression of Beclin-1, LC3-I/II and p62 in the two groups by Western blot (Fig. 4D). It was found that HT22 cells expressed significantly less Beclin-1, LC3-II and more p62 in Aβ1–42 + dioscin + SIRT3 siRNA group (Figs. 4E, F, H), indicating that downregulation of SIRT3 dramatically inhibited autophagy in HT22 cells exposed to Aβ1–42 oligomers even though these cells were pretreated with dioscin. The expression of LC3-I had no significant difference between the two groups (Fig. 4G). Therefore, it was implicated that SIRT3 was an important regulator in dioscin-induced autophagy.

(A) The relative mRNA expression of SIRT3 determined by real-time PCR in HT22 cells treated with NC siRNA or SIRT3 siRNA. (B) The protein band of SIRT3 determined by Western blot. (C) The relative protein expression of SIRT3. (D) The protein bands of Beclin-1, LC3-I, LC3-II and p62 determined by Western blot in HT22 cells treated with Aβ1–42 oligomers, dioscin and NC siRNA (or SIRT3 siRNA). The relative protein expression of (E) Beclin-1, (F) LC3-II, (G) LC3-I and (H) p62. * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with NC siRNA group. + p < 0.05, ++ p < 0.01 and +++ p < 0.001, compared with Aβ1–42 + dioscin + NC siRNA group.
To further investigate whether dioscin protect against Aβ1–42 oligomers-induced neurotoxicity through autophagy, HT22 cells were treated with 3-MA, an autophagy inhibitor. HT22 cells in Aβ1–42 + dioscin + SIRT3 siRNA group and Aβ1–42 + dioscin + 3-MA group showed much lower proliferative activities compared with Aβ1–42 + dioscin + NC siRNA group (Fig. 5A). This result demonstrated that inhibition of autophagy strongly weakened the protective effect of dioscin and exacerbated Aβ1–42 oligomers-induced neurotoxicity. Moreover, downregulation of SIRT3 was also harmful on the protective effect of dioscin. TUNEL staining showed more apoptotic HT22 cells in Aβ1–42 + dioscin + SIRT3 siRNA group and Aβ1–42 + dioscin + 3-MA group (Fig. 5B). Detection of apoptosis and ROS intensity by flow cytometry confirmed that Aβ1–42 + dioscin + SIRT3 siRNA group and Aβ1–42 + dioscin + 3-MA group had significantly increased apoptotic rates, as well as strongly enhanced ROS intensity (Figs. 5C, D, E, F). To avoid potential off-target effects, SIRT3 siRNA-2 was used to verify the functions of SIRT3. As determined by real-time PCR and Western blot, the relative mRNA and protein expression of SIRT3 were effectively downregulated by SIRT3 siRNA-2 (Figs. S1A–C). The result of MTT assay showed that the proliferative activity of HT22 cells was greatly decreased in Aβ1–42 + dioscin + SIRT3 siRNA-2 group (Fig. S1D). Furthermore, the analysis of TUNEL staining and flow cytometry demonstrated that HT22 cells transfected with SIRT3 siRNA-2 exhibited much more apoptotic cells when the cells were treated with dioscin and exposed to Aβ1–42 oligomers (Figs. S1E–G). These results confirmed that dioscin functioned through SIRT3. We then determined the autophagy-related genes of Beclin-1, LC3I/II and p62 by Western blot (Fig. 5G). It was found that HT22 cells transfected with SIRT3 siRNA or treated with 3-MA showed decreased levels of Beclin-1, LC3-II and increased level of p62, comparing to HT22 cells transfected with NC siRNA (Figs. 5H, I, K). Interestingly, the decline or increase of these genes seemed to be more obvious in Aβ1–42 + dioscin + 3-MA group, which implying that SIRT3 was a critical but not the only mediator of autophagy. The expression of LC3-I had no significant difference among the three groups (Fig. 5J). These results indicated that both SIRT3 and autophagy played dominant roles in resisting Aβ1–42 oligomers-induced neurotoxicity.

(A) OD values determined after MTT assay in HT22 cells treated with NC siRNA, SIRT3 siRNA or 3-methylademine (3-MA). (B) Representative images of TUNEL staining. Scale bar = 100 µm. (C) Apoptosis of HT22 cells determined by flow cytometry. (D) Apoptotic rate of HT22 cells. (E) ROS generation determined by flow cytometry. (F) Fluorescence intensity of ROS. (G) The protein bands of Beclin-1, LC3-I, LC3-II and p62 determined by Western blot. The relative protein expression of (H) Beclin-1, (I) LC3-II, (J) LC3-I and (K) p62 in HT22 cells. * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with Aβ1–42 + dioscin + NC siRNA group.
Deposition of Aβ and protein tau has been identified as the characteristic pathology in AD, which damages the neuron and leads to the decline of memory, cognitive function and learning.4) Aβ consists of polypeptides with 40–43 residues of length, including a major proportion of Aβ1–40 (approx. 90%) and a smaller amount of Aβ1–42 (approx. 10%).20) Although the level of Aβ1–40 is much higher than Aβ1–42, growing evidences have confirmed that Aβ1–42 plays a dominant role in forming the plaques in cerebrum since Aβ1–42 is longer and more hydrophobic than Aβ1–40.21–23) Moreover, it is indicated that Aβ1–42 has fast kinetics of aggregation and should be blamed for initiating the pathogenic cascade of AD.24) In the present study, Aβ1–42 oligomers was applied to induce neurotoxicity in HT22 cells. HT22 hippocampal neuronal cell line is an ideal in vitro model to investigate neurodegenerative diseases such as AD and Parkinson’s disease.25) In agreement with previous researches, we found that Aβ1–42 oligomers induced great toxicity, which dramatically inhibited the proliferative activity of HT22 cells. Moreover, Aβ1–42 oligomers induced a large amount of apoptosis and generation of ROS in HT22 cells. These results demonstrated the successfully establishment of in vitro model. Present study aimed to investigate the potential therapeutic effects of dioscin on HT22 cells exposed to Aβ1–42 oligomers.
Dioscin is a natural herb extract with multiple pharmacological activities of anti-inflammation, anti-tumor and anti-oxidation. More recently, researches show that dioscin has neuroprotective effects on cerebral ischemic/reperfusion injury.26) However, little is known about the effect of dioscin on the neurodegenerative disease of AD. In present study, we found that dioscin could decrease Aβ1–42 oligomers-induced neurotoxicity in HT22 cells by enhancing the proliferative activity of these cells in a dose-dependent manner. Furthermore, dioscin could dramatically reduce the apoptotic rate and ROS generation in HT22 cells exposed to Aβ1–42 oligomers. Oxidative stress has been proved to participate in the pathogenesis of AD, which leads to the oxidation of mitochondrial DNA, protein and lipid in brain cells, and eventually the cell apoptosis and death.27) The present study demonstrated the promising protective effects of discin on Aβ1–42 oligomers-induced neuron damages.
SIRT3 is a mitochondrial sirtuin deacetylase, which regulates the mitochondrial proteins and initiates metabolic adaptations to manage ROS generation.28) Moreover, SIRT3 can enhance the ability of antioxidants to scavenge the excessive ROS. Dysregulation of SIRT3 has been reported to be associated with many diseases, including cancer, metabolic diseases and neurodegenerative diseases.29) In a mouse model of AD, the expression of SIRT3 was found to be significantly downregulated, suggesting that SIRT3 might involve in the development of AD.30) Here, we found that the expression of SIRT3 was significantly downregulated in HT22 cell exposed to Aβ1–42 oligomers. However, cells pretreated with dioscin showed greatly enhanced level of SIRT3 even after exposing to Aβ1–42 oligomers. The result indicated that dioscin could upregulate the expression of SIRT3 and SIRT3 might be involved in the neuroprotective pathways, which might partially explain the effects of dioscin on protect against the Aβ1–42 oligomers-induced neurotoxicity.
More interestingly, our study showed that dioscin could induce the autophagy in HT22 cells. Autophagy is a “self-eating” process within the cells, through which the damaged organelles or abnormal aggregated proteins can be degraded by lysosomes.31) Autophagy is an effective approach to clear the aggregated Aβ, prevent the formation of abnormal plaques and maintain the homeostasis of Aβ in normal brains. During the process of autophagy, there are several important genes involved in the cascades. Beclin-1 interacts with the class III phosphatidylinositol-3-kinase (VPS34) and autophagy-related proteins (Atg) to form macromolecular complexes, which generate phosphatidylinositol-3-phosphate that promote autophagosomal membrane nucleation.20) LC3-I and LC3-II are major regulators in autophagy and the conversion from LC3-I to LC3-II is an important indicator of autophagosome formation.32) p62 is a multidomain protein that interacts with autophagy regulator of Atg8/LC3, and then the combined p62 is degraded in autolysosome.33) The occurrence of autophagy results in the degradation of p62 while the deficient of autophagy leads to the aggregation of p62. Unfortunately, emerging evidences indicate that autophagy is inhibited in brain tissues of patients with AD.9) Reduced activity of autophagy is also observed in animal models of AD.34) In our study, much less autophagosomes and autolysosomes were observed in HT22 cells treated with Aβ1–42 oligomers. Besides, the levels of Beclin-1 and LC3-II were noticeable decreased while the level of p62 was enhanced in Aβ1–42 oligomers-treated HT22 cells, confirming that autophagy was inhibited by Aβ1–42 oligomers. Surprisingly, we found that dioscin could induce the upregulation of Beclin-1 and LC3-II, as well as the formation of autophagosome and autolysosome. These results suggested that dioscin could strengthen autophagy in HT22 cells even though they were exposed to Aβ1–42 oligomers.
Although SIRT3 has been reported to be neuroprotective, little is known about the underlying mechanisms. It was found in our study that SIRT3 was involved in the regulation of autophagy since knocking down SIRT3 significantly inhibited the expression of Beclin-1 and LC3-II, as well as led to the aggregation of p62. This finding indicated that SIRT3 played a positive role in promoting autophagy, which might provide a new understanding on the neuroprotective function of SIRT3. Nevertheless, this research just established an association between SIRT3 and autophagy. More studies need to be carried out to investigate the potential signaling pathways. In further study, we found that knocking down SIRT3 or inhibiting autophagy greatly reduced the neuroprotective effect of dioscin on HT22 cells exposed to Aβ1–42 oligomers. Moreover, directly inhibiting autophagy resulted in more dramatical changes of autophagy-related genes. These results indicated that SIRT3 could be an important but not the only regulator in autophagy and dioscin-induced autophagy might be a predominant approach for HT22 cells to resist from the neurotoxicity induced by Aβ1–42 oligomers. Most importantly, it was demonstrated that dioscin could protect against Aβ1–42 oligomers-induced neurotoxicity through regulating the expression of SIRT3 and promoting autophagy, which meant that SIRT3 and autophagy were tightly associated with each other and they contributed together to neuroprotective mechanisms of dioscin.
In summary, dioscin could significantly decrease Aβ1–42 oligomers-induced neurotoxicity in HT22 cells. Dioscin also inhibited apoptosis and ROS generation in HT22 cells exposed to Aβ1–42 oligomers, demonstrating the neuroprotective effect of dioscin. Further study showed that dioscin upregulated the level of neuroprotective SIRT3 and induced autophagy in HT22 cells, revealing the possible mechanisms on the function of dioscin. This study suggested that dioscin might be a promising drug to protect against Aβ1–42 oligomers-induced neurotoxicity and reduced neuron damages or death in AD.
This study was supported by Grants from the Guide Project for Natural Science of Liaoning Province (No. 2019-ZD-0616) and the Doctoral Startup Foundation of Department of Science and Technology, Liaoning Province (No. 201601357).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.