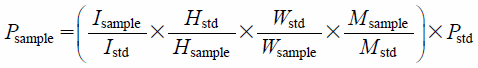Experimental
We recorded 1H- and 13C-NMR spectra using a JEOL ECZ600 (600 MHz) spectrometer. Chemical shifts were reported in delta (δ) units, part per million (ppm) relative to the center of solvent peaks as an internal reference (CDCl3: 7.26 for 1H-NMR, 77.0 for 13C-NMR; CD3OD: 3.30 for 1H-NMR, 49.0 for 13C-NMR; Acetone-d6: 2.04 for 1H-NMR, 29.8 for 13C-NMR). HR-MS were obtained using a Thermo Scientific UltiMate 3000 UHPLC-Q Exactive MS using the electrospray ionization (ESI) method (conditions: column, ACQUITY UPLC HSS T3 (2.1 × 100 mm, 1.7 µm; Waters); mobile phase, A = 0.1% formic acid in H2O, B = 0.1% formic acid in CH3CN; gradient elution, 5–95%B over 5 min; flow rate, 0.4 mL/min). Column chromatography was performed using medium pressure chromatography (Smart Flash, YAMAZEN) equipped with a Hi-Flash column and an Inject column (40 µm). All experiments were performed under anhydrous conditions in an argon atmosphere, unless otherwise mentioned.
5-Bromo-2,4-bis[(2-methoxyethoxy)methoxy]phenol (7)To a solution of 1 (3.2 g, 8.14 mmol) in CH2Cl2 (40 mL), mCPBA was added (approximately 35% water, 2.6 g, 9.77 mmol), and the reaction mixture was stirred at room temperature for 16 h in the presence of Ar gas. The reaction mixture was quenched with saturated aqueous NaHCO3, and then extracted using CH2Cl2 (20 mL × 2). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product was dissolved in CH2Cl2 (40 mL), and 10% aqueous KOH/MeOH (10 mL) was added. After stirring at room temperature for 2 h, the reaction mixture was diluted with EtOAc (100 mL), quenched with 10% HCl, and then extracted with EtOAc (30 mL × 2). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hexane/EtOAc = 30 : 70) to give compound 7 as a colorless oil (2.9 g, 93%).
1H-NMR (600 MHz, CDCl3) δ: 7.13 (s, 1H), 6.97 (s, 1H), 6.47 (s, 1H), 5.22 (s, 2H), 5.21 (s, 2H), 3.90–3.87 (m, 4H), 3.60 (td, J = 4.8, 1.8 Hz, 2H), 3.58 (td, J = 4.8, 1.8 Hz, 2H), 3.41 (s, 3H), 3.38 (s, 3H).
13C-NMR (151 MHz, CDCl3) δ: 147.0, 144.4, 143.3, 119.6, 108.1, 106.7, 96.7, 95.3, 71.5, 71.5, 69.1, 67.9, 59.0 (1C overlapped).
HRMS (ESI) m/z Calcd for C14H25BrNO7+ 398.0809 [M + NH4]+ 398.0806.
{7-[(2-Methoxyethoxy)methoxy]-4-oxo-4H-chromen-3-yl}boronic Acid (8)To a solution of 3 (1.6 g, 4.9 mmol) with anhydrous 1,4-dioxane (25 mL), bis(pinacolato)diboron (3.1 g, 12.0 mmol) was added and degassed by argon bubbling for 10 min. To the above mixture, [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride dichloromethane adduct (198 mg, 0.24 mmol) and AcOK (1.43 g, 14.5 mmol) was added, and the resulting mixture was stirred at 90°C for 6 h. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure, and the residue was dissolved in EtOAc, washed with 1 M HCl and brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to give the crude product 8, which was used in the next reaction without further purification. To determine the structure, the crude product was partially purified by silica gel column chromatography (Hexane/EtOAc = 8 : 2 to 2 : 8) to give 8 as a colorless crystalline solid.
1H-NMR (600 MHz, CDCl3) δ: 8.31 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.14 (d, J = 1.8 Hz, 1H), 7.11 (dd, J = 9.0, 1.8 Hz, 1H), 6.96 (br s, 2H), 3.85 (t, J = 4.5 Hz, 2H), 3.57 (t, J = 4.5 Hz, 2H), 3.38 (s, 3H).
13C-NMR (151 MHz, CDCl3) δ: 182.8, 163.04, 162.0, 158.3, 127.2, 118.3, 116.1, 111.1, 103.5, 93.4, 71.5, 68.3, 59.1.
HRMS (ESI) m/z Calcd for C13H16BO7+ 295.0984 [M + H]+, 295.0981.
5′-Hydroxy-7,2′,4′-tris(2-methoxyethoxymethoxy)isoflavone (5)7)The obtained crude product 8 (4.9 mmol) and 7 (2.2 g, 5.8 mmol) were dissolved in 1,4-dioxane (20 mL) and degassed by argon bubbling under sonication for 10 min. To the above mixture was added [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride dichloromethane adduct (397 mg, 0.49 mmol) and 2 M aqueous K2CO3 (7.3 mL). The reaction mixture was stirred at 100°C for 2 h. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2 and washed with water and brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hexane/Acetone = 65 : 35) to give 5 as a red oil (1.65 g, 62%).
1H-NMR (600 MHz, CD3OD) δ: 8.10 (s, 1H), 8.09 (d, J = 9.0 Hz, 1H), 7.21 (d, J = 1.8 Hz, 1H), 7.15 (dd, J = 9.0, 1.8 Hz, 1H), 7.12 (s, 1H), 6.79 (s, 1H), 5.48 (s, 1H), 5.41 (s, 2H), 5.28 (s, 2H), 5.08 (s, 2H), 3.86 (t, J = 4.8 Hz, 2H), 3.84 (t, J = 4.8 Hz, 2H), 3.69 (t, J = 4.8 Hz, 2H), 3.59 (t, J = 4.8 Hz, 2H), 3.56 (t, J = 4.8 Hz, 2H), 3.48 (t, J = 4.8 Hz, 2H), 3.36 (s, 3H), 3.32 (s, 3H), 3.28 (s, 3H).
7,2′,4′-Tris(2-methoxyethoxymethoxy)-5′-(1,1-dimethylallyloxy)isoflavone (6)A solution of 5 (1.05 g, 1.9 mmol), in anhydrous tetrahydrofuran (25 mL), was degassed by argon bubbling under sonication for 15 min at 0°C. To the above mixture, tetrakis(triphenylphosphine)palladium(0) (110 mg, 0.095 mmol) was added, followed by tert-butyl (2-methylbut-3-en-2-yl) carbonate (1.07 g, 5.7 mmol), and then degassed by argon bubbling under sonication for 5 min at 0°C. The resulting reaction mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with CH2Cl2 and washed with water and brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hexane/Acetone = 70 : 30) to give 5 as a yellow oil (1.05 g, 89%).
1H-NMR (600 MHz, CD3OD) δ: 8.11 (s, 1H), 8.09 (d, J = 9.0 Hz, 1H), 7.22 (d, J = 2.4 Hz, 1H), 7.15 (dd, J = 9.0, 2.4 Hz, 1H), 7.13 (s, 1H), 7.00 (s, 1H), 6.16 (dd, J = 18.0, 10.8 Hz, 1H), 5.41 (s, 2H), 5.27 (s, 2H), 5.16 (dd, J = 10.8, 1.2 Hz, 1H), 5.15 (s, 2H), 5.06 (dd, J = 10.8, 1.2 Hz, 1H), 3.86 (td, J = 4.8, 1.8 Hz, 2H), 3.84 (td, J = 4.8, 1.8 Hz, 2H), 3.71 (td, J = 4.8, 1.8 Hz, 2H), 3.59 (td, J = 4.8, 1.8 Hz, 2H), 3.56 (td, J = 4.8, 1.8 Hz, 2H), 3.49 (td, J = 4.8, 1.8 Hz, 2H), 3.36 (s, 3H), 3.32 (s, 3H), 3.28 (s, 3H), 1.42 (6H, s).
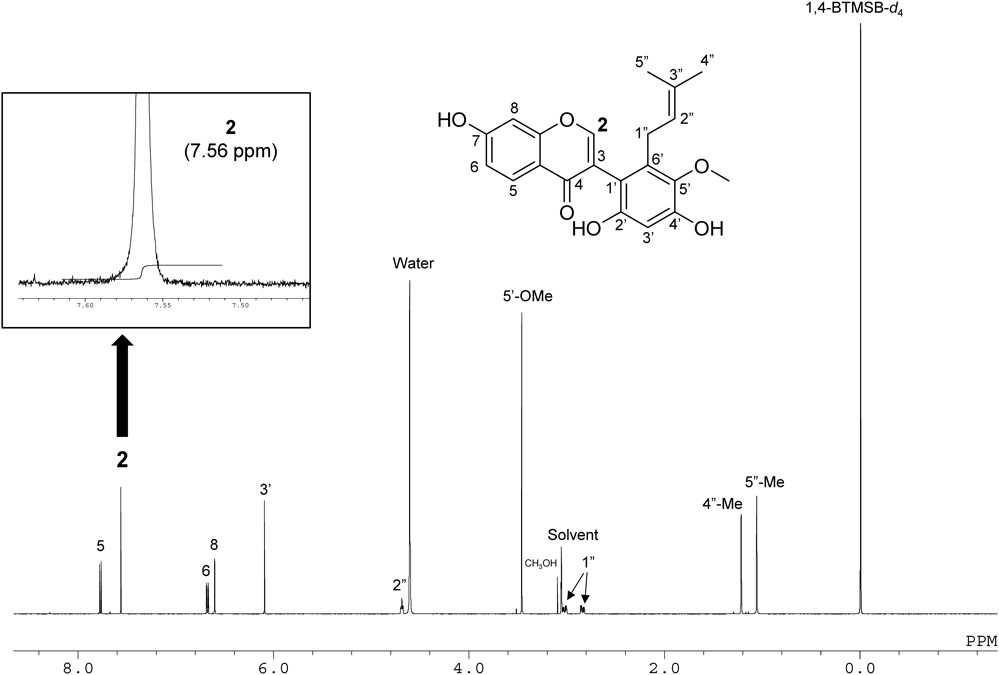
Kwakhurin (KWA)KWA was synthesized from compound 6 according to reported protocols.7)
1H-NMR (600 MHz, Acetone-d6) δ: 7.99 (d, J = 9.0 Hz, 1H), 7.82 (s, 1H), 6.97 (dd, J = 9.0, 2.1 Hz, 1H), 6.90 (d, J = 2.1 Hz, 1H), 6.38 (s, 1H), 4.98 (t, J = 6.9 Hz, 1H), 3.69 (s, 3H), 3.29 (dd, J = 14.5, 6.2 Hz, 1H), 3.07 (dd, J = 14.5, 6.9 Hz, 1H), 1.48 (s, 3H), 1.37 (s, 3H).
13C-NMR (151 MHz, Acetone-d6) δ: 176.4, 163.0, 159.0, 155.7, 153.3, 151.3, 140.1, 136.4, 130.8, 128.3, 124.8, 121.5, 118.8, 115.3, 111.5, 103.1, 102.4, 61.1, 27.5, 25.6, 17.6.
qNMR Measurement1. Reference Standard for qNMR and Solvent
In this study, 1,4-BTMSB-d4 (1,4-bis(trimethylsilyl)benzene-d4, MW = 226.50, Code No. 024-17031, Lot. TWN2900, purity 99.9%), a certified reference material (NMIJ CRM), was purchased from FUJIFILM Wako Pure Chemical Corporation, Ltd. and used as the reference standard for qNMR. Methanol-d4 with the following deuteration rate was used as a solvent for qNMR determination: Lot. A0373436 (100.0 atom%D) by Acros Organics.
2. Instruments and Equipment
The Ultra-Microbalance XPR2UV (Mettler Toledo) with a minimum reading of 0.0001 mg was used. JNM-ECA600 (600 MHz) was used for the NMR. The Wilmad 535-PP-7 was used as an NMR sample tube.
3. Preparation of the Sample Solution
Approximately 3.3 mg of KWA and approximately 1 mg of 1,4-BTMSB-d4 (reference standard for qNMR), which were precisely weighed and placed in the same vial together for each tare, were dissolved in NMR solvent (1 mL). This solution (0.6 mL) was sealed in an NMR sample tube.
4. Conditions for qNMR Determination
The observed spectrum width was 20 ppm. A digital filter was used. The center of the spectrum was set at 5 ppm. The pulse width was set to the time at which a 90-degree pulse was obtained. Acquisition time, 4 s; digital resolution, 0.25 Hz; and delay time, 60 s (The T1 values of the signals of KWA were 0.5 to 4.8 s). An auto FG shim was used for shim adjustment. The determination temperature was set at room temperature (20–30°C). We performed 13C decoupling with MPF8. The dummy scan was performed twice, and the scan was performed 32 times. In principle, the determination was performed 3 times for each sample in accordance with the internal standard method (AQARI: Accurate quantitative NMR with internal reference substance) to ensure that the S/N of the quantitative signal was 200 or higher. Purity Pro, manufactured by JEOL Ltd., was used for NMR data processing. The trimethylsilyl peak of the reference standard for qNMR (1,4-BTMSB-d4) was set at 0 ppm. Phase correction and baseline correction were performed manually. The integration range for each peak was determined using a manual method. All integrated values in this study were expressed in terms of purity (%). The purity of the reagents was calculated using the following formula based on a previous study8–10):
-
I = signal area, H = number of protons, W = weight, M = molecular weight, P = purity (%), std,sample = reference standard for qNMR and sample
The following numbers were used for the calculation: number of protons of methyl groups in 1,4-BTMSB-d4 (reference standard for qNMR), 18; molecular weight of 1,4-BTMSB-d4, 226.50; molecular weight of KWA, 368.38.