Review
Nonheme Iron- and 2-Oxoglutarate-Dependent Dioxygenases in Fungal Meroterpenoid Biosynthesis
2020 Volume 68 Issue 9 Pages 823-831
Details
2020 Volume 68 Issue 9 Pages 823-831
This review summarizes the recent progress in research on the non-heme Fe(II)- and 2-oxoglutarate-dependent dioxygenases, which are involved in the biosynthesis of pharmaceutically important fungal meroterpenoids. This enzyme class activates a selective C–H bond of the substrate and catalyzes a wide range of chemical reactions, from simple hydroxylation to dynamic carbon skeletal rearrangements, thereby significantly contributing to the structural diversification and complexification of the molecules. Structure–function studies of these enzymes provide an excellent platform for the development of useful biocatalysts for synthetic biology to create novel molecules for future drug discovery.
Non-heme Fe(II)- and 2-oxoglutarate-dependent (Fe/2OG) dioxygenases are versatile enzymes for the activation of C–H bonds.1–4) This enzyme family is widely distributed in the primary and secondary metabolism in bacteria, fungi, plants, and vertebrates, and its members accept both small and large molecules to perform a variety of reactions, including simple hydroxylation, epoxidation, desaturation, dealkylation, cyclization, halogenation, and rearrangement reactions. Previous mechanistic and structural studies have established the catalytic mechanism of the Fe/2OG dioxygenases, which utilize mononuclear non-heme Fe(II) and the co-substrate 2OG to couple substrate oxidation to the concomitant decarboxylation of 2OG to yield succinate1–4) (Fig. 1). The resulting active Fe(IV)-oxo species then activates the selective C–H bond of the substrate and abstracts a hydrogen atom to generate a radical species. In most cases, the oxygen rebound completes the enzyme reaction to afford a hydroxylated product. Interestingly, some fungal Fe/2OG dioxygenases are multi-functional and a single enzyme catalyzes several reaction steps, including the remarkable chemistries of dynamic carbon skeletal rearrangement.5–13) This review summarizes our recent progress in research on interesting Fe/2OG dioxygenases involved in the biosynthesis of pharmaceutically important fungal meroterpenoids.14–31)

(Color figure can be accessed in the online version.)
Meroterpenoids are partly terpenoid-derived hybrid natural products with significantly diverse and complex structures (Fig. 2) and include the immunosuppressant drug mycophenolic acid (1) from Penicillium brevicompactum, the potent acyl-CoA: cholesterol acyltransferase (ACA T) inhibitor pyripyropene A (2) from Aspergillus fumigatus, and the promising drug candidate against African trypanosomiasis ascofuranone (3) from Acremonium egyptiacum.9–13) Meroterpenoid biogenesis starts with the synthesis of a polyketide core, and subsequent prenylation, oxidation, and cyclization reactions lead to the formation of the basic skeleton, which is further modified by a variety of tailoring reactions. As a result, significantly complex and diverse structures are produced from simple building blocks in relatively few steps. The biosynthetic pathways include many unusual enzyme reactions with fascinating chemistries.5–13) In particular, Fe/2OG dioxygenases, as well as other oxygenase enzymes, such as heme-dependent CYP monooxygenases and flavin-dependent monooxygenases, significantly contribute to the dramatic increase in the structural diversity and complexity of the molecular scaffolds.

(Color figure can be accessed in the online version.)
AndA, in the biosynthetic pathway of anditomin (4) in Emericella variecolor, is an unusual Fe/2OG dioxygenase that catalyzes sequential oxidations of preandiloid B (8) to construct the characteristic bicyclo[2.2.2]octane ring of andiconin (10)20,27) (Fig. 3). Thus, AndA first installs the conjugated Δ1,2 double bond of preandiloid C (9), which is then converted to andiconin (10) with the complex bridged-ring system. Remarkably, this unprecedented carbon skeletal reconstruction is catalyzed by a single multi-functional enzyme. In particular, the first desaturation reaction is a simple dehydrogenation by an Fe/2OG dioxygenase, whereas the second rearrangement is a highly unusual isomerization reaction catalyzed by an oxidative enzyme. Interestingly, AndA requires ascorbate as a reducing agent in vitro, reflecting the fact that AndA functions as an isomerase in the second reaction.

(Color figure can be accessed in the online version.)
The proposed mechanism of bicyclo[2.2.2]octane ring formation20,27) involves the initial abstraction of the H-12 hydrogen atom of preandiloid C (9) by the active Fe(IV)-oxo species formed by the oxidative decarboxylation of 2OG to generate a C-12 radical (Fig. 3). The subsequent C–O bond cleavage at C-8 yields a C-5′ radical (route A), which is followed by the C–C bond formation between the electron-rich C-12 and the electrophilic C-5′. The following C–C bond formation between the resulting nucleophilic C-8 and the electron-deficient C-2′ generates a radical with the bicyclo[2.2.2]octane ring. Notably, as mentioned above, the AndA enzyme reaction requires ascorbate for the reduction of the radical and Fe(III) to complete the formation of the bridged-ring system.
Computational calculations have supported the proposed mechanism and validated the dynamic conformational changes during the carbon skeletal rearrangement27) (Fig. 4). Actually, there are two possible pathways after the initial formation of the C-12 radical species, depending on the timing of the C–O bond cleavage and the C–C bond formation (Fig. 3). Thus, in route A, as described above, the C–O bond cleavage precedes C–C bond formation, while in route B, C–C bond formation occurs first. Although the calculation suggested that both routes can proceed at room temperature, route A is favored because of its much lower activation energy (ΔG values of all reaction steps are less than 20 kcal/mol), which is consistent with the fact that route B violates Baldwin’s rules. This result also suggested that the primary function of the enzyme is the activation of the selective C–H bond to generate the radical species, and the subsequent rearrangement reaction can proceed without elaborate enzymic assistance, depending on the structures and reactivities of the substrate/intermediates. Nonetheless, it is remarkable that this dynamic multi-step chemistry is catalyzed by a single enzyme.

(Color figure can be accessed in the online version.)
The crystal structure of AndA was solved in the apo form at 2.5 Å resolution, and the complex structures with 2OG and the substrate, preandiloid B (8) or preandiloid C (9), were solved at 2.3 and 2.0 Å resolutions, respectively27) (Fig. 5). As in the other Fe/2OG dioxygenases, AndA maintains the conserved jelly-roll fold (double-stranded β-helical core structure)32) with the Fe-binding 2-His-1-carboxylate facial triad (H135, D137, H213), while 2OG is coordinated to Fe in a bidentate fashion, together with a water molecule and the facial triad, to form an octahedral iron complex. The homodimeric crystal structures revealed that both substrates are loaded on the enzyme in almost identical positions on the lid-like region between the two monomers of AndA, which is formed by loop A and loop B′ from the other monomer. Notably, loop A was only observed when the substrate was bound but not observed in the apo structure, suggesting that it is highly flexible and important for substrate binding. In the complex structures, the D/E-rings of the substrate are tightly fixed by a hydrogen bond network in the lid-like region, whereas the A/B/C-rings are relatively loosely bound to the enzyme by a single hydrogen bond between O1 and N121. On the other hand, the position of the oxo group of 2OG in the active site is thought to be proximal to the H2O molecule coordinated to H135 and to the carboxylic oxygen of 2OG bound to H213.

(Color figure can be accessed in the online version.)
Structure-based site-directed mutagenesis confirmed the importance of the residues lining the active site of the enzyme.27) Interestingly, the substitutions of residues E66, N121, R239, and Y272′, which are involved in substrate binding, revealed that these residues are not crucial for the first desaturation reaction but are essential for the second bridged-ring formation, suggesting that a precisely organized active site architecture is required for the bicyclo[2.2.2]octane ring formation. Considering the fact that the ring reconstruction takes place on the B/C/D rings, the hydrogen bonds of O-3/O-5 on the D ring with I70 and Y272′ via an H2O molecule seem to be important, while the hydrogen bonds with the A-ring are relatively distant from the reaction center.
Interestingly, the final step of anditomin biosynthesis in E. variecolor is catalyzed by AndF, another Fe/2OG dioxygenase sharing 36% amino acid sequence identity with AndA.20) Thus, AndF catalyzes further oxidative rearrangement to produce the highly congested ring structure of anditomin (4) (Fig. 6). In this case, the reaction is initiated by abstracting the H-11 hydrogen atom of andilesin C (11) with the seven-membered lactone A-ring to yield a C-11 radical species, which is followed by hydroxylation to yield the Fe(II)-bound intermediate. Subsequently, the elimination of the hydroxy group and the concomitant 1,2-acyl shift with H-9′ deprotonation would generate the end product anditomin (4). To further investigate the structure–function relationships of the Fe/2OG dioxygenases, biochemical and biophysical characterizations of AndF are now in progress in our laboratories.

(Color figure can be accessed in the online version.)
In the biosynthesis of austinol (5) in Aspergillus nidulans, the multifunctional Fe/2OG dioxygenase AusE, sharing 38% identity with AndA, constructs the characteristic spiro-ring in two consecutive reactions19,33) (Fig. 7). The first reaction is the desaturation of preaustinoid A1 (12) with the seven-membered lactone A-ring to produce preaustinoid A2 (13) with a conjugated Δ1,2 double bond, which is again accepted by AusE as a substrate to undergo the second reaction, generating the spiro-lactone preaustinoid A3 (14) by an unprecedented carbon skeletal rearrangement.19) Notably, this is the first report describing the oxidative formation of a spiro-carbocycle structure by an Fe/2OG dioxygenase. In this case, in the second reaction, the active Fe(IV)-oxo species generated at the active site of the enzyme is proposed to abstract the H-5 hydrogen atom of preaustinoid A2 (13) to form a C-5 radical (Fig. 8A), which is further converted to a cyclopropylcarbinyl C-2 radical intermediate via homoallyl–homoallyl radical rearrangement and C-1/C-5 bond formation. Finally, the C-1/C-10 bond cleavage and H-9 proton abstraction generate preaustinoid A3 (14), the spiro-lactone with the conjugated Δ9,10 double bond.
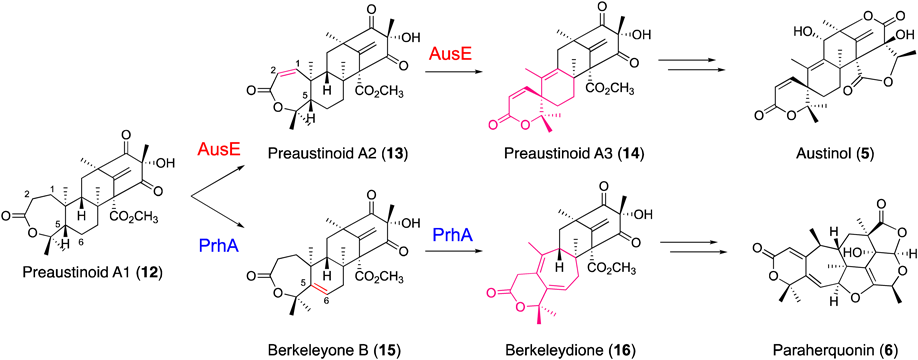
AusE and PrhA use preaustinoid A1 as a common substrate to catalyze divergent multistep oxidation reactions in austinol and paraherquinonin biosynthetic pathways. (Color figure can be accessed in the online version.)

(Color figure can be accessed in the online version.)
Intriguingly, in the upstream biosynthetic pathway of austinol (5), AusE also catalyzes the C-3 dehydrogenation of berkeleyone A (17) to produce the C-3 carbonyl preaustinoid A (18)19) (Fig. 9). Although dehydrogenation is a common reaction in Fe/2OG dioxygenases, it is interesting that AusE, a single enzyme, catalyzes the biosynthetic steps “before” and “after” the formation of the 7-membered lactone A-ring by the flavin-dependent Baeyer–Villiger monooxygenase AusC. Further in vivo and in vitro analyses of the enzyme reactions revealed that AusE also catalyzes the hydroxylation of berkeleyone A (17) to produce 5-hydroxyberkeleyone A (19). Moreover, AusE also accepts preaustinoid A (18) as a substrate to catalyze sequential desaturation and spiro-ring-forming reactions, to generate preaustinoid C (20) with a conjugated Δ1,2 double bond and austinoid C (21) with a spiro-cyclopentenone structure, which is also thought to be formed by an initial H-5 proton abstraction and subsequent skeletal rearrangement. The substrate promiscuity and multi-talented catalytic versatility of the Fe/2OG dioxygenases are remarkable.

(Color figure can be accessed in the online version.)
The X-ray crystal structures of AusE complexed with Mn(II) and 2OG were solved at 2.1–2.8 Å resolution.26) The overall folding of the homo-dimeric AusE is similar to those of other fungal Fe/2OG dioxygenases, including the aforementioned AndA, the endoperoxide-forming FtmOx1 from A. fumigatus in diketopiperazine derivative biosynthesis,34) and the multifunctional AsqJ from A. nidulans in quinolone antibiotic biosynthesis,35,36) despite their low amino acid sequence identity (29–30%). The AusE structure revealed a tight dimer interface (approx. 1500 Å2), which forms a funnel-like active site in the jelly-roll fold with the conserved iron-binding 2-His-1-carboxylate facial triad (H130, D132, H214). The structural comparison with the cycloheptadiene-forming Fe/2OG dioxygenase PrhA and their functional conversions are described below.
PrhA from Penicillium brasilianum, which is highly homologous (78% identity) to the spiro-ring-forming A. nidulans AusE, is another remarkable Fe/2OG dioxygenase that catalyzes a ring-expansion reaction in the biosynthesis of paraherquonin (6), a highly oxygenated and congested hexacyclic fungal meroterpenoid.22) Interestingly, PrhA and AusE share preaustinoid A1 (12) as a common substrate; however, in contrast to AusE that produces preaustinoid A3 (14) with the A-ring spiro-lactone, PrhA catalyzes oxidative B-ring expansion of the substrate to yield berkeleydione (16), with a unique cycloheptadiene structure through two consecutive reactions (Fig. 7). The first PrhA reaction is the installation of the Δ5,6 double bond of berkeleyone B (15), in contrast to AusE, which inserts the conjugated Δ1,2 double bond. Thus, the active Fe(IV)-oxo species abstracts the H-5 proton instead of H-1 of preaustinoid A1. In the following reaction (Fig. 8B), PrhA abstracts the H-1 proton of berkeleyone B (15) to yield a C-1 radical, which is further converted to a cyclopropylcarbinyl radical intermediate via homoallyl–homoallyl radical rearrangement and C-1/C-5 bond formation. Finally, C-5/C-10 bond cleavage and H-1 proton abstraction lead to the construction of the B-ring-expanded cycloheptadiene scaffold of berkeleydione (16). Other examples of ring expansion reactions catalyzed by fungal Fe/2OG dioxygenases include the biosyntheses of the tropolone stipitatic acid37) and the cephalosporin of penicillin.38–40)
It is particularly interesting that the highly homologous PrhA and AusE (78% identity) catalyze conversions of the common substrate preaustinoid A1 into totally different products, the B-ring-expanded berkeleydione (16) and the spiro-lactone preaustinoid A3 (14), respectively (Fig. 7). To investigate the structure–function relationship, X-ray crystal structures of PrhA in the apo-form, in complex with Fe(II), in complex with Fe(II) and 2OG, and in complex with Fe(II), 2OG, and the substrate preaustinoid A1 (12), were solved at 2.1–2.3 Å resolution.26) Comparisons with the above described crystal structures of AusE revealed that the overall structures and active sites of both enzymes are strikingly similar, with the conserved iron-binding 2-His-1-carboxylate facial triad (Fig. 10). Notably, in the PrhA complex structure with the substrate, the A-ring of preaustinoid A1 (12) is fixed in a boat, instead of a chair, conformation, upon binding to the active site, which would be the reason why the initial proton abstraction takes place at H-5 instead of H-1 of the substrate by the Fe(IV)-oxo species. The structures also revealed the dynamic conformational change for loop A (residues 57–69), from an open state to a closed form, upon substrate binding. Interestingly, in the complex structure, the D-ring of preaustinoid A1 (12) is in close contact with 2OG suggesting that 2OG binds to the active site prior to the substrate loading.

(A) The active site is separated into three regions. Region L includes the 2-His-1-Asp triad binding Fe(II). Region B includes the residues interacting with the D-ring of 12. Region R includes residues surrounding the A/B-ring of 12. (B) Comparison of AusE-Mn/2OG and PrhA-Fe/2OG/12 active sites. (Color figure can be accessed in the online version.)
Close examination of the PrhA and AusE structures revealed that almost all of the residues lining the active site are conserved, and the only exceptions are residues 150, 232, and 241 (V150, A232, and M241 in PrhA; L150, S232, and V241 in AusE) in the region surrounding the A-ring of the common substrate preaustinoid A1 (12)26) (Fig. 10B). Substitutions of these three residues with the corresponding amino acids in the other enzyme successfully resulted in the functional interconversion of PrhA and AusE. Thus, both triple mutants completely lost their original activity and instead gained the function of the other enzyme with almost equal catalytic efficiency. In addition, the PrhA V150L/A232S/M241V mutant exhibited further expanded catalytic functions, and this single enzyme now efficiently catalyzes five consecutive oxidation reactions to generate a variety of products, including unnatural novel meroterpenoids 22–25 (Fig. 9). It is remarkable that such dramatic changes in the substrate and product selectivity can be achieved by simple mutations of a few residues lining the active site cavity.
To further investigate the molecular basis for the expanded catalytic functions, the crystal structures of the mutants in complex with Fe(II), 2OG, and the four distinct substrates, 12, 13, 14, or 17, were solved at 2.3 Å resolution26) (Fig. 11). These structures revealed that all four substrates bind to the active site in almost identical positions, and the common D-ring region of the substrates is tightly fixed by the same hydrogen bonding network, whereas the A/B-ring region of the substrates is bound to the active site with few interactions. Because of this loose molecular recognition, the enzyme could accept substrates with variable A/B-ring structures and perform the unusual multiple oxidation reactions.

(Color figure can be accessed in the online version.)
NvfI from Aspergillus novofumigatus is another unusual Fe/2OG dioxygenase that forms an endoperoxide in the biosynthesis of novofumigatonin (7), a highly oxygenated meroterpenoid with a unique orthoester structure.28) Remarkably, NvfI installs “three” oxygen atoms in a single enzyme reaction and accepts asnovolin A (26) as a substrate to produce fumigatonoid A (27) containing an endoperoxide linkage between C-13 and C-2′ (Fig. 12). The proposed reaction mechanism involves the initial abstraction of the H-13 hydrogen atom of the substrate by the Fe(IV)-oxo species to generate a C-13 radical intermediate. The subsequent incorporation of molecular oxygen yields a peroxy radical, without O–O bond scission, which is followed by C–O bond formation at C-2′ to form the C-3′ radical. Finally, NvfI completes the enzyme reaction by the oxygen rebound at C-3′ to install a hydroxy group. This mechanism is similar to that proposed for FtmOx1,34) which is another example of an Fe/2OG dioxygenase that generates an endoperoxide in the biosynthesis of the diketopiperazine-derived natural product verruculogen in Aspergillus fumigatus. However, the radical quenching mechanism of FtmOx1 is different from that of NvfI in that it requires a reducing agent as a hydrogen donor to complete the reaction, resulting in the incorporation of only “two” oxygen atoms into the substrate.28)

(Color figure can be accessed in the online version.)
Many endoperoxide-containing natural products have been isolated and shown to exhibit significant biological activities. The endoperoxide-forming reaction catalyzed by the Fe/2OG dioxygenase NvfI is also similar to that by the well-characterized prostaglandin endoperoxide H synthase (PGHS), although it is totally different because PGHS is a heme-containing enzyme.28) Except for PGHS in prostaglandin biosynthesis, the Fe/2OG dioxygenase FtmOx is the only known enzyme that produces an endoperoxide. Interestingly, NvfI and FtmOx share only 17% amino acid sequence identity, and further structure–function analyses of these rare endoperoxide-forming enzymes are in progress in our laboratories.
The fungal meroterpenoid biosynthetic pathways exhibit an abundance of unusual chemistries and interesting enzyme reactions, including those of the multi-functional Fe/2OG dioxygenases that perform selective C–H activation/functionalization with unusual substrate promiscuity and catalytic versatility. This intriguing chemistry is governed by the inherent structures and reactivities of the substrates and molecular recognition by the enzymes. The chemical reactions take place in the relatively unreactive environment of the active site of the enzyme, which protects the active Fe(IV)-oxo species and the generated radical intermediates from undesired side reactions. The catalytic versatility of the Fe/2OG dioxygenases is also derived from the plasticity of the ligands surrounding the Fe-binding center. In addition, 2OG, substrates, and hydrogen bond networks with the active site residues are crucial for fine-tuning the activity of each enzyme. Further elucidation of the structure–function relationships of these enzymes will reveal more detailed molecular bases for their fascinating chemistries, and provide an excellent platform for the development of versatile biocatalysts to supply novel molecules for drug discovery.
The author would like to express sincere appreciation to an excellent group of coworkers whose contributions are cited in the text. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (Japan Society for the Promotion of Science KAKENHI Grant Nos. 22108004, 23241068, JP15H01836, JP16H06443 and JP20H00490).
The author declares no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2019 Pharmaceutical Society of Japan Award.