Abstract
The biologically active, naturally occurring 1,2,3,4-tetrahydroisoquinoline-quinone (THIQ) family members isolated from Actinomycetes and marine organisms have been studied thoroughly over the past five decades. Among them, marine natural products along with their reduced compounds, such as renieramycins and ecteinascidins, have attracted interest due to their fantastic structures and meager availability in nature as well as their potent antitumor profiles. As part of our search for new anticancer metabolites through the isolation and characterization of anticancer THIQ compounds from Thai marine animals, we have developed a fascinating THIQ natural product chemistry and medicinal chemistry based on knowledge of the chemistry of saframycin antibiotics as well as their isolation, characterization, transformation, partial synthesis, and total synthesis. This review mainly presents our contributions during 1999–2019 to the field of research on biologically active renieramycin along with ecteinascidin marine natural products.
Introduction
Marine species provide enticing sources of potential new pharmaceutical agents. An additional incentive is the lure of fascinating drug targets from marine sources. A high percentage of marine-derived natural products have novel structures and consequently identify new pharmacophores for structure–activity relationship studies (SARs).1,2) Unfortunately, some marine natural products are available only in very minute quantities. Therefore, it has been very difficult to obtain a consistent supply of target compounds for development by isolation from marine organisms or by total synthesis and because severe limitations on the amounts of natural products from marine organisms have thus far precluded detailed biological evaluation.
In 1969, the first example of a naturally occurring 1,2,3,4-tetrahydroisoquinolinequinone (THIQ) family member was the crude aqueous extract of the colonial tunicate Ecteinascidia turbinate, which was reported to possess in vivo antitumor activity.3) A number of research groups attempted to isolate the active constituents in the extract and determine their chemical structures. However, the isolation and structural determination of the family members was not completed until 1990 by Professor Rinehart’s group at Illinois University.4) The rigid pentacyclic core system was a well-known chemotype originally reported from microbes, where the compound classes were saframycins, naphthyridinomycins, and quinocarcins.5,6) Additionally, it connected to the third isoquinoline group through a 10-membered lactone ring together with a spiro system.4,7–9) Many specialists in the field of natural product chemistry were very interested in discovering similar families from marine sponges and their associated nudibranchs, such as renieramycins and cribrostatin 4 (Fig. 1).
In this review, some examples of biologically active THIQ marine natural product chemistry research conducted in our laboratory, including isolation, structure elucidation, chemical transformation, total synthesis, and SAR studies are introduced.
1. Chemistry of Saframycin Antitumor THIQ Natural Products: Research Background
We have studied the chemistry of saframycin antibiotics since 1980 and succeeded not only in the isolation and structure elucidation of some minor components (such as saframycins D–H as shown in Fig. 2) but also in the second total synthesis of 5b.6) During our research, we found several useful proofs as described below.
1.1. Structure Elucidation of Saframycins with a Single 1,2,3,4-Tetrahydro-isoquonoine-5,8-dione in the Basic Core SystemWe succeeded in preparing the two isomers 13 and 14 from 12 in the final step of 5b10) (Chart 1).

Chart 1. Conversion of Pentacyclic Compound 12 into 5b
In the 1H-NMR spectra of 5b and 13, diagnostic homoallylic coupling (J value = 2–3 Hz) between 1-H and 4-Hß through five bonds was observed. On the other hand, this coupling was negligible in the spectra of 12 and 14 along with those of any pentacyclic synthetic intermediates of 5b (Fig. 3). These observations clearly indicate that the terminal p-quinone ring is oriented toward the left side. High-resolution (HR) MS also confirmed this conclusion.11)
1.2. Selective Oxidation at the C14 Position to Confirm a Plausible Biosynthetic PathwayWe are particularly interested in the structure of saframycin family members, because the left-terminal ring of all saframycins is in the p-quinone form alone, but their right-terminal rings exhibit a variety of oxidation levels, such as phenols, p-quinones, and hydroquinones. Thus, we constructed our working hypothesis for the interconversion reactions of saframycins as shown in Chart 2. It involves the initial oxidation of a type I compound (such as a safacin) to form a type II compound (such as saframycins A and B), into which a hydroxyl group might be introduced at the C14 position to give a type III compound (such as saframycin G), which in turn would be oxidized and reduced (redox) to afford a type IV compound (such as saframycins D and F). On the other hand, the introduction of a methoxy group at the C14 position of a type II compound would give a type V compound (such as saframycin C), which could then be reduced to a type VI compound (such as saframycin Mxs).12)

Chart 2. Working Hypothesis on the Interconversion of Saframycin Family Members
The challenge addressed in this transformation was the stereoselective introduction of a hydroxyl group into the C14 position of the pentacyclic framework region. A preliminary demonstration of the above transformation was carried out using the tricyclic model compound 15.13) Thus, we successfully prepared 15 through two differing pathways, as shown in Charts 3A and 3B.

Chart 3A. First-Generation Preparation of Phenol 15

Chart 3B. Practical 15-Step Synthesis of Phenol 15
The first synthesis of phenol 15 started from the readily available tricyclic lactam 1614,15) (Chart 3A). Partial demethylation of 16 with BBr3 (2 equiv.) in CH2Cl2 at 0 °C for 1 h gave 17 in 72% yield, the structure of which was confirmed by X-ray analysis. Acylation of 17 with trifluoromethanesulfonic anhydride (Tf2O) and trimethylamine (TEA) in CH2Cl2 at –20 °C for 30 min afforded 18 in 87% yield. Following the Orter procedure,16) treatment of 18 with formic acid, TEA, palladium acetate, and 1,1-bis(diphenylphosphino)ferrocene (DPPF) in dimethylformamide (DMF) at 60 °C for 2 h gave 19 in 89% yield. Selective demethylation of 19 with sodium benzylselenoate,17) which was prepared in situ by the reaction of excess sodium borohydride and dibenzyl diselenide in DMF under reflux for 2 h, gave the desired phenol 15 and its isomer 20 in 28 and 17% yields, respectively, along with a 38% recovered yield of 19.
An improved 15-step synthesis of 15 as a right-ring model from the 3-hydroxy-4-methoxy-5-methylbenzandehyde derivative 2118) was achieved, as shown in Chart 3B.19) Condensation of 21 with 1,4-diacetylpiperazine-2,5-dione (22) in the presence of potassium tert-butoxide in DMF gave 3-arylidenepiperazine-2,5-dione (23) in 74% yield following the procedure of Gallina and Liberatori.20,21) The secondary amine of 23 was protected with the 4-methoxybenzyl group to give 24, which was subjected to successive treatment with hydrazine hydrate followed by methylation to afford 25 in 74% overall yield. Deprotection of 25 with concentrated H2SO4 and trifluoroacetic acid (TFA) (1 : 20) and then reduction of the double bond with hydrogen over 20% palladium hydroxide on carbon in EtOH accompanied by debromination gave 26 in 75% overall yield. The O-benzylation of 26 with benzyl bromide and K2CO3 in DMF and then introduction of the amide nitrogen with isopropyl chloroformate and 4-dimethylaminopyridine (DMAP) gave 27 in 89% yield. Debenzylation of 27 followed by bromination afforded 28 in 83% yield. Chemoselective reduction of 28 with an excess of lithium tri-tert-butoxyaluminohydride in THF followed by cyclization with TFA gave 29 in 97% yield. Finally, conversion of 29 into 15 was finished in three-step sequences (84%).
With the phenol model 15 in hand, we examined how it could conform to the proposed biosynthetic process, as shown in Chart 2.22) Oxidation of 15 with bis(salicidene)ethylidene)ethylene-diiminocobalt(II) (salcomine)23) in DMF at 25 °C for 5 h afforded the p-quinone 30 in 64% yield. It was identical in all respects to an authentic sample, which was obtained by 10N HNO3 oxidation of 17 in 87% yield (Chart 4). The key step was the introduction of an oxygenated functional group into the C14 position of the tricyclic model 17. In the chemistry of tetra-substituted p-benzoquinones, duroquinone and 2,3-dimethyl-1,4-naphthoquinone are known to react with a variety of nucleophiles such as enolates or amines to produce side-chain oxidation products.24) As C14 was at an allylic positon, we succeeded in introducing an α-hydroxy group using selenium oxide (SiO2). Treatment of 30 with 1.1 equiv. of SeO2 in dioxane under reflux for 4 h gave 31 in 80% yield stereoselectively. The hydroxyl stereochemistry of 31 was assigned on the basis of 1.0-Hz coupling between H-13 and H-14. Furthermore, the reaction of 30 with SeO2 in MeOH under reflux for 30 h gave 32 in 61% yield. Then, treating 30 with SeO2 (2 equiv.) in p-xylene produced the ketone 33 and the hydroquinone 34 in 7 and 30% yields, respectively. Although catalytic reduction of p-quinone 30 (H2, 10% Pd/C, in EtOAc) seemed to occur quantitatively as judged by TLC detection, the catalyst was removed by filtration and washed with EtOAc, and the combined colorless filtrates were concentration in vacuo to give a pale yellow solid 30 (86% recovered), probably because the initiating hydroquinone converted 30 by air oxidation. In contrast, hydrogenation of 31 and 32 under the same conditions resulted in the isolation of hydroquinones 35 (69%) and 36 (61%), respectively. These results confirmed the high stability of hydroquinone forms bearing a hydroxyl and methoxy group at C14.

Encouraged by the results of tricyclic model studies, we successfully applied the syntheses of saframycins C–E from saframycin B (5b)22,25) (Chart 5A). As the saframycin pentacyclic ring system has the potential to form similar benzylic methylene carbon atoms at the C4 and C14 positions, a regiochemical problem might occur during the oxidation reaction. We were, however, able to predict that SeO2 oxidation might be highly selective due to the difference in the steric environment at the C4 and C14 carbon atoms. Thus, treatment of (±)-5b with 2 equiv. of SeO2 in dioxane at 25 °C for 72 h afforded (±)-5d (15.6%) along with the hydroxyl compounds 36 and 37 in 40.0 and 4.5% yields, respectively (10.9% yield of 5a was recovered). Reaction of (±)-5b with 2.25 equiv. of SeO2 in MeOH at 25 °C for 88 h gave (±)-5c and 36 in 44.7 and 19.1% yield, respectively. Although some spectroscopic data on saframycin E (5e) were reported by Professor Arai’s group in 1977, its structure could not be established due to its instability.26) Fortunately, we found 100-MHz 1H-NMR spectral data together with electron ionization (EI) MS, IR, and UV data on triacetate 38, which was obtained by the reaction of 5e with acetic anhydride in pyridine in 50% yield. Thus, hydrogenation of 36 with 10% Pd/C in EtOAc gave the leuco compound 39, and then treating 39 with silica gel in EtOAc in the presence of oxygen gas gave synthetic 5e in 62.4% yield. As direct comparisons of the synthetic 5e with the natural one have not been possible, we prepared triacetate 38 (56.1%). The synthetic 38 was clearly identical to the triacetyl derivative of 5e by comparisons of UV, IR, MS, and 1H-NMR data as well as TLC behavior.

Chart 5A. Preparation of (±)-Saframycins C–E from (±)-Saframycin B and Structure Determination of Saframycin E
We also succeeded in converting saframycin A into F via saframycin G, as shown in Chart 5B.25,27)

Chart 5B. Transformation of Saframycin A to F via Saframycin G
In this project, we had to resolve two additional problems. The first was that no observable reaction occurred when saframycin A (5a) was reacted with SeO2 in dioxane. This problem was resolved by using acetic acid as a solvent to give saframycin G (5g; 30.4% yield) along with saframycin G acetate 40 (7.1%), saframycin F (5f, 3.8%), and 14-epi-saframycin G 41 (7.9%). The second problem was that the yield of the transformation of 31 into 34 with SeO2 in p-xylene under reflux was only 30%. Accordingly, it might better to convert 31 into 34 in two-step sequences. Encouraged by the results of model studies, oxidation of 5g with Dess–Martin periodinate (DMP)28) in CH2Cl2 at 25 °C for 7 h gave ketone 42 in 75% yield. After hydrogenation of 42 with 10% Pd/C in EtOAc, the dark yellow reaction mixture became colorless, indicating that the bis-hydroquinone might be produced. During workup, however, selective air oxidation occurred in the terminal left ring to produce saframycin F (5f) in 73% overall yield.
2. Chemistry of THIQ Marine Natural Products
2.1. Isolation and Structure of THIQ Marine Natural Products (1982–2000)Renieramycins A–D were isolated from a bright blue sponge, Reniera sp., found near Isla Grande, Mexico, by Frincke and Faulkner in 1982.29) Interestingly, the pentacyclic core ring system of renieramycins was the same that of saframycins, but the relative stereochemistry was epimeric at the C1 position, based on the difference nuclear Overhauser effect (NOE) experiments. Several years later, He and Faulkner reported the isolation of very unstable renieramycins E (9e) and F (9f) from the blue sponge Reniera sp., both of which were collected from a marine lake in Palau, western Caroline Islands. At that stage, they pointed out that the C1S stereochemistry of 9a–d as published should be revised into C1R based on careful NOE studies30) (see Fig. 1). At almost the same time, Fukuyama et al. completed an elegant first total synthesis of renieramycin A (9a) and independently reached the same conclusion on the C1R stereochemistry.31) In 1992, renieramycin G (9g) was isolated from the Fijian blue sponge Xestospongia caycedoi, and the structure of 9g was deduced by Davidson from its spectral data.32) Since 9g was the first example in which all of the carbon signals were assigned on the bases of the 1H- and 13C-NMR spectral data through 1H-detected heteronuclear multiple-bond coherence (HMBC) experiments, it confirmed that both the detailed 1H-NMR spectral analysis of THIQ family members and 13C-NMR spectral analysis are useful methods for structure determination.
However, there are many problems, especially in the field of marine natural products, because of the very small amounts of substances available from marine organisms and of the inherent instability of the molecules. Paramenswaran et al. reported a new type of THIQ renieramycin H (43), which was isolated from the methanol extract of the bright blue sponge Haliclona cribricutus along with renieramycin I (9i)33) (Fig. 4). Renieramycin H (43) has a novel structure and was the first example of a dimeric THIQ natural product having a hydroxyl group at the bridgehead C13 position.
During the course of our synthetic studies of the right-side model of saframycins such as phenol 15, we completed the synthesis of the tricyclic ring model compound 4434) from easily available 4512,13) in six steps, as shown in Chart 6. In this project, we found a regioselective hydroxylation of 45 by metal–hydrogen interchange, and subsequent reaction of the organometallic intermediate with nitrobenzene afforded 46 in 53% yield.13) The introduction of a nitro group at the C18 position of 46, followed by acetylation and reduction sequencing, gave the aniline derivative 47 in 76% overall yield. Oxidation of 47 with potassium nitrosodisulfonate (Fremy’s salt) at 25 °C for 1 h gave p-quinone 48 in 84% yield. Treatment of 48 with 1N HCl in MeOH under reflux for 40 h gave 44 and 49 in 42 and 36% yields, respectively. Surprisingly, heating 49 with 1N HCl in MeOH for 5 d afforded 44 in 82% yield. The probable mechanistic pathway for the introduction of the methoxy group at the C14 position of 49 is shown in Chart 7.35)

Chart 6. Preparation of Compound 44 from 45 in Six Steps

Chart 7. Mechanism of Conversion of 49 into 44 via a Hydroquinone Intermediate
In the case of an original report of the structure of renieramycin H (43), for which the 13C-NMR carbon signals were assigned as shown in Fig. 5 except that two carbonyl carbon signals were missing (at C18 and C24). In this report, a carbon signal at δ 108.0 ppm was assigned to the novel bridgehead C13 carbon of structure 43. The unusual chemical shift at the C13 carbon signal could not be readily explained, but we suspect it is an aminal carbon connected to an amide carbonyl. As we have the complete 13C-NMR spectral data on compound 44 through a series of HMBC experiments, the quaternary carbon signal at C13 appears at δ 85.6 ppm. Particularly convincing evidence is found in the comparison of the two hydroxyl proton chemical shifts (δ 5.70 ppm and δ 11.34 ppm) with the hydroquinone 34 (δ 5.58 ppm and δ 11.52 ppm). These results indicated that renieramycin H is not a bis-THIQ. In addition, our estimation was supported by comparison of 13C-NMR spectral assignments of the six carbon signals at the right-side terminal arene ring along with the adjacent ketone carbonyl carbon signal at the C14 position of 34 (δ 109.8 ppm). Thus, the structure of renieramycin H (43) should be revised to 9h. Although it was difficult to confirm the stereochemistry at the C1 position of 9h at this stage, Pettit et al. discovered cribrostatin 4 from the blue sponge Cribrochalina sp. collected in reef passages in the Republic of Maldives, the structure of which was determined X-ray crystallographic analysis.36) The NMR spectra of renieramycin H and cribrostatin 4 were identical, and thus the structure of renieramycin H is now assigned as 9h.
In 2000, the Fontana group discovered jorumycin (50) in very minute quantities from the mantle and mucus of the Pacific nudibranch (India), Jorunna funebris (Mollusca: Nudibranchia: Doridina: Kentrodoridae).37) The structure of 50 was elucidated on the grounds of MS data and extensive 2D-NMR analysis (Fig. 6). That was the first example of renieramycin marine natural products having an acetyl ester side chain.
3. THIQ Marine Natural Products from Thai Marine Organisms
Marine species are an enticing source of new pharmaceutical agents with wonderful potential. Marine species also offer an additional incentive as the lure of fascinating fresh drug targets. As early as 2000, some marine-derived natural products were shown to possess novel structures from which new pharmacophores have been identified for SAR studies, but almost all marine natural products including THIQ compounds have lower stabilization within only trace amounts. Thus, they have precluded detailed biological evaluation and few examples have reached the clinical trial stage.
In 1999, we started an international cooperative research project entitled “Medicinal Chemistry on Alkaloids from Plants in the Malaya Area” supported by the Japan Society for the Promotion of Science (JSPS). As this project developed with specialists in Thailand, Malaysia, and Indonesia, we were fortunate to meet an ongoing key person, Dr. Khanit Suwanborirux of the Faculty of Pharmaceutical Sciences, Chulalongkorn University, Thailand. He reported the following exciting information on Thai THIQ marine natural products.
3.1. Isolation and Structure of the Stabilized Renieramycin-Type Derivatives, Renieramycin M, Obtained on Gram Scale from the Thai Blue Sponge Xestospongia sp.Dr. Suwanborirux had already collected a Thai blue sponge Xestospongia sp. in the vicinity of Sichang Island, Gulf of Thailand, at a depth of 3–5 m in July 1992. Extraction was performed on 15 kg of wet animal, followed by LH-20 column chromatography and preparative reversed-phase (C18) chromatography, resulting in the isolation of renieramycins J (9j), K (9k), and L (9l), of which the structures along with relative stereochemistry were elucidated on the basis of individual spectroscopic analysis38) (Fig. 7).
It is interesting that these renieramycins have a propan-2-one unit at the C21 position. One possible pathway to generate these compounds is the partial reduction of the lactam carbonyl (type A, such as renieramycin G [9g]) to afford the carbinolamine (type B, such as renieramycin E [9e]), into which an enol nucleophile of propan-2-one can be introduced at the C21 position to give the Mannich adduct (type D, such as 9j]) via the iminium cation species (type C) stereoselectively (Chart 8).

Following our working hypothesis, we reported the model transformation from 30 to 54, as shown in Chart 9.39) Reduction of the lactam 3022) with 65% toluene solution of sodium bis-(2-methoxyethoxy)aluminium hydride (Red-Al) in THF at 0 °C for 1 h yielded the leuco compound 51. After the reaction mixture was quenched with AcOH, an aqueous solution of KCN was added dropwise over 10 min, and this mixture was stirred at 25 °C for 1 h to give a mixture of 52 and 53, which in turn was subjected to oxidation with 60% HNO3 to give 53 in 74% overall yield from 30. Finally, acid-catalyzed Mannich-type reaction of 53 with 4 equiv. of silver nitrate (AgNO3) in acetone at 60 °C for 2 h gave 54 in 31% yield. An NOE between 14-Hß (δ 2.23, d) and 21-H (δ 3.14, dd) revealed the relative stereochemistry at C21-Hß) of 54.

Chart 9. Preparation of Renieramycins K and L by the Mannich Reaction
In this stage, although we were excited to discover a new source of renieramycins from Xestospongia sp. in Thailand, it was difficult to rule out the possibility that renieramycins J–L were artifacts resulting from propan-2-one exchange during the separation and purification steps. These results indicate that the precursor of 9j should be designated as renieramycin E (9e), which has a carbinolamine group that may be relatively unstable during the purification and isolation procedure.
In the preliminary chemistry of saframycin antibiotics, in 1980 Arai and his group isolated and characterized a labile minor product, saframycin S (5s in Fig. 1) having a hydroxyl group at the C21 position.40) Saframycin S (5s) was transformed into saframycin A (5a) by NaCN in phosphate buffer in 60% yield. The reverse process from 5a into 5s was also possible by the reaction of 5a in 0.1 N H2SO4 at 120 °C for 40 min in 41% yield (Chart 10). Saframycins present an inherent difficulty in the development of satellite antibiotics for chemotherapeutic agents because of their extremely low production in culture, but Arai et al. succeeded after the unexpected observation that cyanide was directly incorporated into the cyano group of saframycin A (2a).40,41) These findings might help to equalize the labile THIQ compounds with a hemiacetal function at the C21 position and its reproduction, especially in THIQ marine natural products.

Chart 10. Interconversion Reactions of Saframycins A (5a) and S (5s)
Based on the above findings, we attempted to stabilize renieramycin E (9e) by conversion into homogenized renieramycin M (9m) by the addition of KCN to the homogenized sponge before starting workup. Thus, the Suwanborirux team used scuba divers to collect Xestospongia sp. (8.4 kg wet weight) in the vicinity of Sichang Island at a depth of 3–5 m in December 2001 and then prepared the frozen marine animals. The samples were subjected to a solvent partition to give a crude fraction (16.2 g), trituration of which with MeOH afforded the precipitate of 9m. Silica gel chromatography of the filtrate gave renieramycins M (9m, 1.822 g) and N (9n, 247 mg) (Fig. 8A). Renieramycin M (9m) was easily transformed into renieramycin E (9e) by treatment with AgNO3 in 69% yield. It showed data identical to those of an authentic standard in all respects.30) Renieramycin N (1n) was sensitive to light and air oxygen as well as to pH above 7. It was rapidly oxidized to give the corresponding bis-p-quinone (named renieramycin O: 9o).42)
The isolation of renieramycins using the above procedure is advantageous because it provides for a practical way to isolate and purify THIQ-type labile marine natural products. We continued investigating the remaining extracts and obtained four minor compounds named renieramycins O (9o), Q (9q), R (9r), and S (9s).43) Furthermore, we discovered new renieramycins T (9t) and U (9u), of which the structures were the first examples of fascinating ecteinascidin–renieramycin hybrid marine natural products.44) In addition, from the low polar fraction, we found renieramycin V (9v),45) which has a sterol moiety attached to renieramycin M (9m) (Fig. 8B).
3.2. Isolation and Structure of Renieramycins from the Thai Nudibranch Jorunna funebrisThe nudibranch J. funebris (Mollusca: Gastropoda: Opisthobranchia: Nudibranchia: Kentrodorididae) is a marine, slug-like invertebrate belonging to the phylum Mollusca which lacks a protective hard shell.46) J. funebris (20 animals; 500 g wet weight) which was feeding on the blue Xestospongia sp., was collected in the vicinity of Sichang Island at a depth of 3–5 m in March 2004 and its egg ribbons (23.2 g wet weight) were separately obtained in November 2004. The animals were carefully dissected into two parts, the mantles and visceral organs (combined digestive glands and gonads). Then, all three parts, including the egg ribbons, were separately homogenized with phosphate buffer (pH 7). Aqueous KCN solution was added to each suspension, the mixture was macerated with methanol, and then the extract was filtered. A general extraction procedure of each part gave the residue (mantle, 700 mg; visceral organs, 980 mg; egg ribbons, 270 mg). Each residue was separately subjected to flash column chromatography on silica gel to give THIQ-type compounds, as summarized in Table 1. We succeeded in isolating three new compounds, jorunnamycins A–C (55a–c) along with four known compounds, renieramycins M (9m), N (9n), O (9o), and Q (2q)47) (Fig. 9).
Table 1. THIQ Marine Natural Products from Aqueous KCN-Pretreated
J. funebris. | Mantle 700 mg | Visceral organs 980 mg | Egg ribbons 270 mg |
|---|
| New compounds | | | |
| Jorunnamycin A (55a) | 7.5 | ND | 3.8 |
| Jorunnamycin B (55b) | ND | 23.6 | ND |
| Jorunnamycin C (55c) | ND | ND | 21.7 |
| Known compounds | | | |
| Renieramycin M (9m) | 25.3 | ND | 62.1 |
| Renieramycin N (9n) | 0.5 | 9.2 | NT |
| Renieramycin O (9o) | 6.4 | 2.0 | NT |
| Renieramycin Q (9q) | ND | 1.3 | ND |
As we wanted to establish sustainable collaborative relationships to expand research on common biologically active marine natural products to other countries, we contacted the specialist Professor G.P. Concepcion of the University of the Philippines through the JSPS Asia and Africa Science Platform Program for assistance in 2010–2020 projects.48) We found three new THIQ-type marine natural products, renieramycins W (9w), X (9x), and Y (9y), along with two known renieramycins M (9m) and T (9t) from the Philippine blue sponge Xestospongia sp. growing in the vicinity of Puerto Galera, Oriental Mindoro, Mindoro Island49) (Fig. 10). It appeared that it might be a better source for an approximately 3-fold increase in the yield of 9m compared with the yield from the Thai blue sponge. Furthermore, both 9w and 9x are the first examples of tiglic acid ester derivatives, which are geometrical isomers at the C1 unsaturated ester of known angelate derivatives 1m and 1t, respectively. In addition, we also found that 9y is the first example having a penta-substituted phenol at the terminal ring of the left side in renieramycin-type marine natural products.
3.3.2. Structure of Renieramycin P and Funnebricins A and BFusetani et al. reported the isolation and structure elucidation of renieramycin P (9p)50) from the organic extract of the Japanese violaceous sponge Neopetrosia sp. collected off Kuchinoerabu Island, Satsunan Archipelago, at a depth of 20 m in July 2001. The structure of 9p including the absolute stereochemistry was elucidated by stereoscopic and chemical conversion51) (Fig. 11). The cytotoxicity of 9p against 3Y1, HeLa, and P388 cells (IC50, nM) were 5.3, 12.3, and 0.53, respectively.
In 2014, Guo and his group succeeded in isolating fennebricins A (56a) and B (56b) from the acetone extracts of the skin of the South China Sea nudibranch J. funebris and its possible sponge-prey Xestospongia sp., but the yields of both compounds were minute and their biological activities could not be tested.52) Two years later, Guo reisolated 56a along with new fennebricins C (56c) and D (56d) to take a different sample collected in Ximao Island, Hainan Province, P. R. China53) (Fig. 11). Preliminary bioassay results and SARs suggested that a few isolated compounds were potential to be drug reads.
4. Total Synthesis of Renieramycin-Type Marine Natural Products
Before 2000, Fukuyama performed pioneering total syntheses in the field of biologically active THIQ natural products, i.e., saframycins A (5a),54) B (5b),55,56) and renieramycin A (9a).57) In addition, we prepared 21-dehydroxyjorumycin (57) and 21-decyanojorunnamycin C (58) from the key intermediate 5910) in our saframycin B synthesis58) (Chart 11). Even though both compounds were analogues of natural products, it was gratifying to confirm our prediction more than 10 years previously that small amounts of acetyl or propanoly ester derivatives would be present in the renieramycin marine natural products jorumycin (50)37) and jorunnamycin C (55c).47)

Chart 11. Preparation of Renieramycin Derivatives 57 and 58
Many biochemists are interested in renieramycin-type marine natural products with antineoplastic activity, but it is difficult to determine whether this group compound has promising antitumor activity because of its scarcity from natural sources excluding total synthesis. The results of partial models studied including the chemistry of saframycin antibiotics described above and the availability of renieramycin M (9m) have enabled us to prepare and evaluate new renieramycin-type compounds.
According to the model conversion by the Mannich-type reaction from 53 to 54 as shown in Chart 8, treatment of 9m with AgNO3 in acetone at 50 °C for 1 h gave 9j in 70% yield.59) After converting saframycin G (5g) into F (5f) via compound 42 in two steps as shown in Chart 5B, we reported the transformation of renieramycin O (9o) into Q (9q) by DMP oxidation and catalytic reduction along with an air oxidation process27) (Chart 12).

Chart 12. Transformation of Renieramycin M (9m) into J (9j) and Renieramycin O (9o) into Q (9q)
We also reported the successful transformation of 9m into jorumycin (50) via jorunnamycin A (55a), as shown in Chart 13.59) The crucial step in this process involves the removal of the angeloyl ester group of 9m. Numerous attempts to remove the ester group from the angalate side chain of 9m under hydrolytic conditions were unsuccessful because it has vinylogous esters in both p-quinone rings. In contrast, the reduction of 9m with aluminum hydride (AlH3) in THF at 0 °C for 1 h followed by air oxidation gave jorunnamycin A (55a) with a maximum yield of only 26%. Accordingly, hydrogenation of 9m with 20% Pd/C in EtOAc for 3 h gave the leuco compound 64, which was subsequently treated with 8 equiv. of AlH3 in THF at –20 °C for 4 h followed by air oxidation to provide 55a in 53% yield along with the C21-decyanated compound 62 (14%) and the recovered 9m (21%). Acetylation of 55a with acetyl chloride, TEA, and DMAP in CH2Cl2 gave the acetate 65 in 73% yield. Finally, treatment of 65 with AgNO3 in aqueous MeCN at 40 °C for 2 h afforded jorumycin (50) in 83% yield. As it has been one of the universal synthetic targets of biologically active THIQ marine natural products, to date, one semisynthesis of 50 from safracin B (6B) in six steps (24% overall yield)60) and five total syntheses of 50 have been achieved.61–65) All strategies to synthesize a characteristic pentacyclic core are based on electrophilic substitution of the electron-rich arenes, such as the modified Pictet–Spengler cyclization.66)

Chart 13. Transformation of Renieramycin M into Jorumycin via Jorunnamycin A
We isolated renieramycin M (9m) in gram scale from the Thai blue sponge described above and thus had access to a variety of renieramycin derivatives for the investigation of SARs, as described below. To extend our investigation of biological activities in the series of renieramycin marine natural products, we are also interested in the structures of renieramycin G (9g) and cribrostatin 4 (=renieramycin H: 9h), which have a lactam carbonyl residue at C21, because both compounds maintained their cytotoxicity despite the lack of the essential functional group of hemiaminal or acetonitrile at C21. We completed the total synthesis of renieramycin G and cribrostatin 4.
4.3.1. Total Synthesis of (±)-Renieramycin GFive total syntheses of renieramycin G have been reported,61,62,65,67,68) and in 2011, we reported our first total synthesis of (±)-renieramycin G (9g) from readily available 2-hydroxy-3-methyl-4,5-dimethoxybenzaldehyde (66) in 25 steps (1.0% overall yield).69) One year later, we completed its improved total synthesis from 66 in 21 steps (6.3% overall yield), as shown in Charts 14A and 14B.70)

Chart 14A. Total Synthesis of (±)-Renieramycin G (Part 1): Construction of the Key Intermediate

Chart 14B. Total Synthesis of (±)-Renieramycin G (Part 2)
Condensation of the methoxymethyl-protected benzaldehyde 67, which was obtained from 66 in quantitative yield, with 22 and base gave compound 68 in 73% yield. Catalytic hydrogenation of 68 over 5% Rh/C in 2-propanol at 25 °C for 1 h, followed by introduction of the 2-propyloxycarbonyl group, afforded 69 in 96% yield. Condensation of 69 with 70 and a base gave 71 in 70% yield. The sequence of transformations of 71 into 73a without the purification of intermediates was found to be the best choice in terms overall yield (73a, 83% and 73b, 9%).
Catalytic hydrogenation of 73b through the action of hydrogen (28 atm) on 20% Pd(OH)2/C in EtOH at 80 °C for 41 h from the less hindered α-face followed by detosylation gave 74 in 74% yield. The next stage of the investigation involved the construction of the pentacyclic framework along with the stereochemistry of the side chain (Chart 14B).
After extensive investigation of the reaction conditions based on the results of the modified Avendaño procedure71,72) the following procedure was found to be optimum in terms of product yield and reproducibility of the reaction. The reaction of amine 74 with ethyl diethoxyacetate in the presence of trimethylsilyl trifruoromethanesulfonate (TMSOTf) in CH2Cl2 at 120 °C for 13 h gave cyclized ester 75 in 77% yield. X-Ray crystallographic analysis of 75 revealed that the stereochemistry at the C1 position was epimeric to that of naturel renieramycins.
With the pentacyclic compound in hand, the remaining problem was the isomerization at C1 of 75. Because 75 was not sufficiently soluble in any solvent, it was converted into the corresponding benzyl ester and then transformed in three steps to give the aldehyde 76a (71% overall yield) having the smallest carbonyl functional group at C1. Treatment of 76a with 1.0 equiv. of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in THF at 25 °C for 24 h achieved equilibrium through the enolate 77 to afford 76b and 76a in 62 and 33% yields, respectively. Isomerization of the recovered 76a repeated twice finally gave 76b and 76a in 85 and 4% yields, respectively. Reduction of 76b with sodium cyanoborohydride (NaBH3CN) in THF in the presence of catalytic AcOH gave 78 in 65% yield. Treatment of 78 with angeloyl chloride in CH2Cl2 at 25 °C for 2 h gave the ester 79 in 93% yield. Oxidative demethylation of 79 with ceric ammonium nitrate in aqueous acetonitrile at 0 °C for 15 min afforded (±)-renieramycin G (9g) in 80% yield. Synthetic (±)-9g was shown to be identical to the natural one based on a comparison of their spectroscopic data.
4.3.2. Total Synthesis of (±)-Cribrostatin 4 (Renieramycin H)To date, three total syntheses of cribrostatin 4 have been reported,73–75) and all included the construction of a bicyclic AB ring system with a cis relationship at the C1 and C3 positions, followed by the condensation of the E-ring part and elaboration of the central CD ring. Following the method used for the total synthesis of renieramycin G, we succeeded in a 19-step total synthesis of (±)-cribrostatin 4 along with (±)-renieramycin I (9i) from the compound 80,21) which was the starting material for our saframycin synthesis, in 9.5% overall yield, as shown in Charts 15A–C.
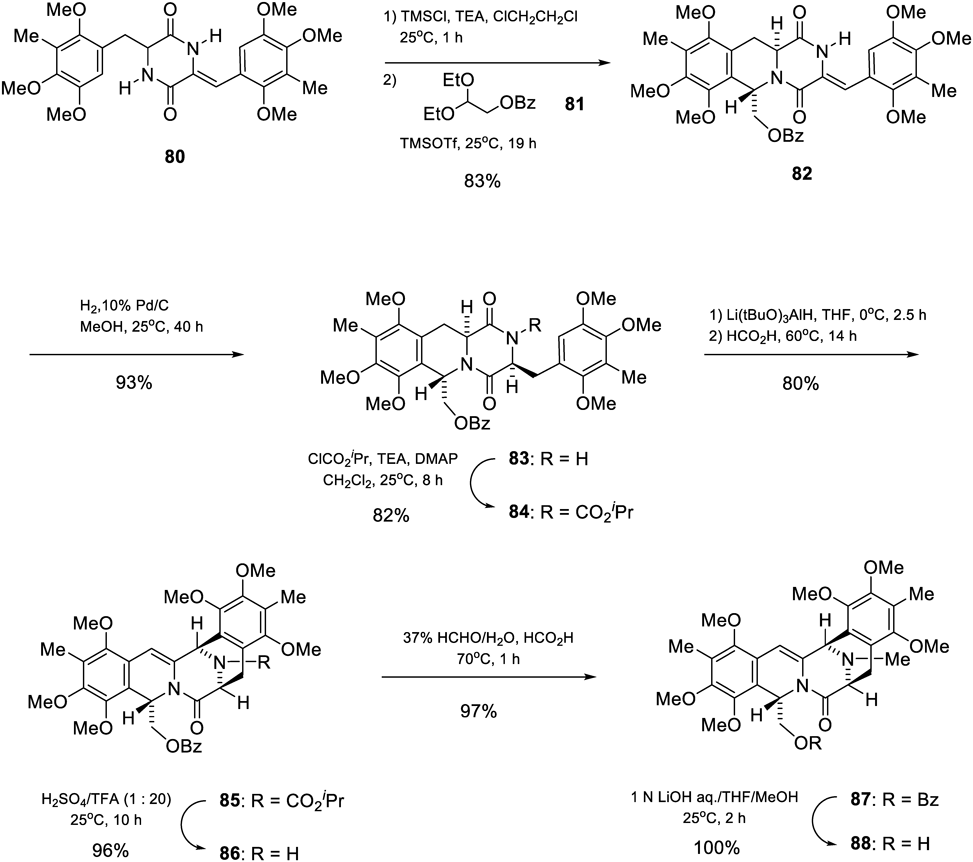
Chart 15A. Total Synthesis of (±)-Cribrostatin 4 (Part 1)

Chart 15B. Plausible Mechanism of Acid-Catalyzed Cyclization Including Isomerization and an Unexpected Oxidation Sequence

Chart 15C. Total Synthesis of (±)-Cribrostatin 4 (Part 2)
Two-step cyclization of 80 with 2,2-diethoxyethyl benzoate (81)76)via the O-trimethylsilyllactim intermediate gave 82 in 83% yield (Chart 15A). As X-ray crystallographic analysis of 82 confirmed that the stereochemistry was trans between the C1 and C3 protons, cyclization might proceed from the less hindered α-face to the (E)-iminium isomer.
The next stage of our investigation involved the establishment of a method to construct the cis stereochemistry between C3 and C13. Catalytic reduction of 82 gave 83 in 93% yield, and the introduction of a 2-propyloxycarbonyl group into 83 afforded imide 84 in 82% yield. Partial reduction of 83 followed by cyclization under acidic conditions gave 85 in 80% yield. Surprisingly, it was an unexpected dehydrogenation product. The mechanistic pathway for this transformation was unclear, but it could be presumed that the dehydration of the aminal (I) to generate an iminium ion species (II) and rapid isomerization (III→VI), followed by a spontaneous oxidation and cyclization sequence generated 8577) (Chart 15B). The deprotection of 85 with TFA and H2SO4 gave 86 (96%), the stereochemical structure of which was confirmed by X-ray crystallographic analysis. Reductive methylation of 86 generated 87 in 97% yield. Hydrolysis of 87 with base afforded 88 in quantitative yield.78)
Following the procedure of our total synthesis of 9g as shown in Chart 14B, four-step transformation of 88 gave the bis-p-quinone 91,74) which was the key intermediate of Williams cribrostatin 4 synthesis79,80) (Chart 15C). Reaction of 91 with angeloyl chloride in CH2Cl2 and DMF afforded 92 in 84% yield. Oxidation of 92 with SeO2 in aqueous dioxane at 80 °C for 6 h proceeded through the chemo- and diastereoselective introduction of a hydroxyl group at C14 to produce 93 in 71% yield. The spectroscopic properties of the synthetic sample were in complete agreement with those of natural renieramycin I (9i).33) Treatment of 93 with 10.5 equiv. of DMP in CH2Cl2 at 25 °C for 3 h, followed by sodium thiosulfate reduction and selective air oxidation of the resulting bis-hydroquinone (leuco compound), afforded cribrostatin 4 (=renieramycin H: 9h) in 84% overall yield.
5. Isolation of Ecteinascidin 770 from the Thai Tunicate Ecteinascidia thurstoni
5.1. Discovery of the Thai Tunicate and Isolation of Ecteinascidins 770 and 786The isolation and structure elucidation of ecteinascidin marine natural products from the Caribbean tunicate Ecteinascidia turbinate were reported by Rinehart et al. in 1990–1996.4,81–84) Among this group, ecteinascidin 743 (10a) was demonstrated to possess extremely potent in vitro cytotoxicity in a variety of tumor cell lines.85) Ecteinascidin 743 (10a) has the international nonproprietary name trabectedin and is being developed under the trade name Yondelis. It was approved in 2007 for use in humans with soft tissue sarcoma in the EU and is currently being used in over 18 countries and regions around the world, including the U.S.A. and Japan. Despite its medicinal importance, there are few SAR studies on trabectedin owing to the meager amount of the natural product available and the low chemical diversity of the biosynthetically produced derivatives.84,86)
Thus, its novel structure, combined with meager availability from nature and unique mechanism of action, has made 10a an attractive and important synthetic target. To date, four total syntheses have been achieved by the groups of Corey,87,88) Fukuyama,89,90) Zhu,91) and Ma.92) Formal total syntheses were also reported by the groups of Danishefsky93) and Williams.94) Based on Corey’s excellent total synthesis, a semisynthetic process starting from 21-cyanosafracin B (6c), an antibiotic produced by the fermentation of Pseudomonas fluorescens,95,96) has been developed by Pharma Mar in the synthesis of 10a.97,98) This process could take 21 steps for 1.0% overall yield as an industrial route. Recently, Zhang’s group has developed a method for concise, practical semisynthesis of 10a from 6c in 14 steps with 1.5% overall yield.60)
In March 2000, we used scuba divers to collect a Thai tunicate, Ecteinascidia thurstoni Herdman 1891,99) growing around Phuket Island. It was the initial example of an Asian tunicate containing ecteinascidins, and the preliminary attempts to extract the alkaloid were performed on 50 g of wet animal using the Rinehart protocol. However, there were only trace amounts of the alkaloid in the crude fraction. We assumed that any natural products with an α-aminal functionality such as 10a were relatively unstable during extraction. Following the stabilization procedures used for the transformation of saframycin S (5s) and renieramycin E (9e) into saframycin A (5a) and renieramycin M (9m), we isolated ecteinascidin 770 (10d) on gram scale along with ecteinascidin 786 (10e) from the Thai tunicate E. thurstoni after pretreatment with KCN in buffer solution (Fig. 12). The availability of 10d enabled us to prepare ecteinascidin analogues100) promising increased biological activity and a broadened spectrum, as discussed below.
5.2. Reaction of Ecteinascidin 770 and Its DerivativesThe advantage of increasing the yield of labile marine natural products enabled us to prepare novel ecteinascidin derivatives with improved cytotoxicity profiles. First, 10d was easily transformed with AgNO3 in aqueous acetonitrile into ecteinascidin 743 (10a) in 97% yield, which was identical to the authentic spectral data in all respects100) (Chart 16). Oxidation of 10d with m-chloroperbenzoic acid (m-CPBA) in CH2Cl2 gave ecteinascidin 786 (10e) in 92% yield.101) Reduction of 10e was performed with phenylsilane/oxo-rhenium complex102,103) in THF at 25 °C for 9 h to generate 10d and 94 in 55.1% and 12.7%, respectively.104) While the reaction of 10d under the same conditions was relatively slow, after continuing for 118 h, 94 was produced in 33.6% yield along with 43.9% of recovered 10d.

Chart 16. Reaction of Ecteinascidin 770 and Its Derivatives
6. Evaluation of Cytotoxicity Profiles of THIQ Marine Natural Products
6.1. Cytotoxicity Profiles of Renieramycin Marine Natural ProductsWith access to a variety of renieramycin marine natural products, we were then able to test them for cytotoxicity against HCT116 human colon carcinoma and QG50 human lung carcinoma cells lines, as shown in Table 2. For the substituent at the C21 position, a cyano or hydroxyl group is essential, and any lactam carbonyl such as 9g, 9h, and 9i as well as the alkylated compound 9j dramatically decreased cytotoxicity. These results suggested that the elimination of the cyano or hydroxyl groups under physiological conditions results in a reactive iminium species that is responsible for covalent bond formation with the drug target, which is similar to the medicinal chemistry findings on saframycin antibiotics.5)
Table 2. Cytotoxicity of Renieramycins and Related Compounds (IC
50 nM)
| Compound | | Cell line | Ref. |
|---|
| HCT116 | QG56 |
|---|
| Renieramycin E | (–) 9e | 0.38 > | 1.0 | 59 |
| Renieramycin G* | (±) 9g | >1000 | >1000 | 70 |
| Renieramycin J | (–) 9j | 730.0 | 510.0 | 59 |
| Renieramycin M | (–) 9m | 7.9 | 19.0 | 42 |
| Renieramycin N | (–) 9n | 5.6 | 11.0 | 42 |
| Renieramycin O | (–) 9o | 28.0 | 40.0 | 42 |
| Renieramycin Q | (–) 9q | 59.0 | 71.0 | 43 |
| Renieramycin R | (–) 9r | 23.0 | 29.0 | 43 |
| Renieramycin S | (–) 9s | 15.0 | 26.0 | 43 |
| Renieramycin V | (–) 9v | >1000 | >1000 | 45 |
| Jorumycin* | (–) 50 | 0.57 | 0.76 | 59 |
| Cribrostatin 4* | (±) 9h | >1000 | >1000 | 79 |
| Renieramycin I* | (±) 9i | 300.0 | 900.0 | 79 |
| 21-CN-jorumycin* | (–) 65 | 0.38 > | 0.68 | 59 |
| Jorunnamycin A | (–) 55a | 13.0 | 59.0 | 47 |
| Jorunnamycin B | (+) 55b | 455.0 | 618.0 | 47 |
| Jorunnamycin C | (–) 55c | 1.5 | 19.0 | 47 |
*Unnatural synthetic product.
The preliminary biological evaluation described above established the basis for conducting new investigations of this class of antitumor marine alkaloids focusing on the effects of variation of the ester analogues at C22.105,106) A total of 43 ester analogues of renieramycin derivatives were prepared from jorunnamycin A (5a) under a variety of conditions in moderate yields. Among them, 4-pyridinecarbonyl derivative 95 showed the most potent cytotoxicity profile, exhibiting 21-fold and 5-fold increases in cytotoxicity to H292 (1.1 ± 0.1 nM) and H460 (1.6 ± 0.3 nM) human non-small cell lung cancer (NSCLC) cell lines, respectively, relative to 9m (Fig. 13). While we found several dramatic developments in the preparation of synthetic analogues such as phthalascidin (96),107) QAD (97),108) and zalypsis (98),109,110) along with their antiproliferative activities, our findings show that the potent antitumor activity of ester analogue 95 may be promising and further efforts to explore the therapeutic potential of other renieramycin analogues are under way.
6.2. Cytotoxicity Profiles of Ecteinascidin Marine Natural ProductsAs a result of continuing efforts to establish the medicinal chemistry of marine-derived antitumor ecteinascidin 743 (10a), we reported the preparation of 17 6′-O-acyl derivatives 99a–q and determined their cytotoxicity against human tumor cell lines HCT116, QG56, and DU145101) (Fig. 14). The nitrogen-containing heterocyclic ester derivatives 99l–q showed in vitro cytotoxicity similar to that of 10d, whereas the other derivatives were less cytotoxic than 10d.
In an attempt to improve cytotoxicity profiles, we next prepared the 31 2″-N-amide analogues 100a–ff in a three-step protocol: 1) 18,6′-O-biallyl protection leading to II; 2) 2′-N-amide formation to III′; and 3) deprotection of the allyl group to generate I104,106,111) (Chart 17).

Chart 17. Preparation of a Variety of 2′-N-Acyl Derivatives of Ecteinascidin 770
The protection of both 18- and 6′-phenolic hydroxyl groups resulted in markedly diminished cytotoxicity, whereas the introduction of acyl groups at the 2′-N position enhanced cytotoxicity. We found that the 2′-N-4-fluorocinnamoyl derivative 100cc had approximately 70-, 13-, and 40-fold greater cytotoxicity to the HCT116, QG56, and DU145 cell lines, respectively, relative to 10d (Fig. 15). These findings indicate that the 2′-N-acyl derivatives play an important role in the antitumor activity of ecteinascidin analogues.
Conclusion
This review introduced our contributions to research on the antitumor renieramycin and ecteinascidin THIQ marine natural products during 1999–2019 at Meiji Pharmaceutical University. We had many opportunities to learn about the elegance and efficiency of the assembly of novel natural molecules along with some accidental proofs and unpredictable positive results. Numerous renieramycin and renieramycin analogues have recently become accessible due to synthetic efforts in several laboratories. We believe that, in combination with the results of recent clinical trials, biosynthetic studies112) and practical syntheses of THIQ marine natural products113) will contribute to applications in drug discovery and development in the near future. While it will be very difficult to predict the final outcomes of such drug discovery efforts, it is clear that a focused approach and combined efforts will accelerate the efficient discovery and development of new marine product-based antitumor agents. We hope that our efforts will inspire the creation of all-Asian medicinal chemistry research teams to develop new anticancer agents based on THIQ marine natural products in the future.
Acknowledgments
These studies were carried out at the Department of Pharmaceutical Chemistry, Meiji Pharmaceutical University (MPU). I would like to thank the staff and students of this laboratory, particularly Drs. Masashi Yokoya and Shinya Kimura. I would also like to express my sincere appreciation to Professor Emeritus Akinori Kubo of MPU for his encouragement in this research field. Furthermore, I appreciate the warm cooperation extended by Dr. Khanit Suwanborirux (Chulalongkorn University), an indispensable partner in Thai marine natural product chemistry research. These studies were mainly supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (Nos. 1659119, 15K07873, and 18K06561) and the JSPS Asia and Africa Science Platform Program (2006–2008, 2010–2012). I dedicate this review to Emeritus Professor Shin-ichiro Sakai (Chiba University, Japan), who passed away on November 17, 2019, for his outstanding contributions to nitrogen-containing biologically active natural product chemistry.
Conflict of Interest
The author declares no conflict of interest.
Notes
This review of the author’s work was written by the author upon receiving the 2019 Pharmaceutical Society of Japan Award for Divisional Scientific Contribution.
References and Notes
- 1) Newman D. J., Cragg G. M., J. Nat. Prod., 67, 1216–1238 (2004).
- 2) Molinski T. F., Dalisay D. S., Lievens S. L., Saludes J. P., Nat. Rev. Drug Discov., 8, 69–85 (2009).
- 3) Sigel M. M., Welham L. L., Lichter W., Dudeck L. E., Gargus J. L., Lucas A. H., “Food Drugs from the Sea Proceedings,” ed. by Younghen H. W. Jr., Marine Technology Society, Washington, D. C., 1969, pp. 281–284.
- 4) Rinehart K., Med. Res. Rev., 20, 1–27 (2000).
- 5) Arai T., Kubo A., “The Alkaloids,” Vol. 21, ed. by Brossi A., Academic Press, New York, 1983, pp. 55–100.
- 6) Kubo A., Saito N., “Studies in Natural Products Chemistry,” Vol. 10, ed. by Atta-ur-Rahman, Elsevier, Amsterdam, 1992, pp. 77–145.
- 7) Scott J. D., Williams R. M., Chem. Rev., 102, 1669–1730 (2002).
- 8) Avendaňo C., la Cuesta E., Curr. Org. Synth., 6, 143–168 (2009).
- 9) Le V. H., Inai M., Williams R. M., Kan T., Nat. Prod. Rep., 32, 328–347 (2015).
- 10) Kubo A., Saito N., Yamato H., Matsubuchi K., Nakamura M., J. Org. Chem., 53, 4295–4310 (1988).
- 11) Kubo A., Saito N., Kitahara Y., Takahashi K., Yazawa K., Arai T., Chem. Pharm. Bull., 35, 440–442 (1987).
- 12) Saito N., Obara Y., Azumaya M., Kubo A., Chem. Pharm. Bull., 40, 2620–2626 (1992).
- 13) Saito N., Obara Y., Aihara T., Harada S., Shida Y., Kubo A., Tetrahedron, 50, 3915–3928 (1994).
- 14) Kurikara H., Mishima H., Tetrahedron Lett., 23, 3639–3640 (1982).
- 15) Kubo A., Saito N., Yamato H., Yamauchi R., Hiruma K., Inoue S., Chem. Pharm. Bull., 36, 2607–2614 (1988).
- 16) Cacchi S., Ciattini P. G., Morera E., Ortar G., Tetrahedron Lett., 27, 5541–5544 (1986).
- 17) Ahmad R., Saa J. M., Cava M. P., J. Org. Chem., 42, 1228–1230 (1977).
- 18) Saito N., Tachi M., Seki R., Sugawara Y., Takeuchi E., Kubo A., Synth. Commun., 30, 2407–2421 (2000).
- 19) Saito N., Tachi M., Seki R., Kamayachi H., Kubo A., Chem. Pharm. Bull., 48, 1549–1557 (2000).
- 20) Gallina C., Liberatori A., Tetrahedron, 30, 667–670 (1974).
- 21) Kubo A., Saito N., Yamato H., Kawakami Y., Chem. Pharm. Bull., 35, 2525–2532 (1987).
- 22) Saito N., Ohira Y., Wada N., Kubo A., Tetrahedron, 46, 7711–7728 (1990).
- 23) Yoshida R., Nakajima S., Ohnuma T., Ban Y., Shibasaki M., Aoe K., Date T., J. Org. Chem., 53, 5355–5359 (1988).
- 24) Findy K. T., “The Chemistry of the Quinonoid Compounds,” Vol. 2, ed. by Patai S., John Wiley and Sons, New York, 1974, pp. 877–1144.
- 25) Saito N., Harada S., Nishida M., Inouye I., Kubo A., Chem. Pharm. Bull., 43, 777–782 (1995).
- 26) Arai T., Takahashi K., Kubo A., J. Antibiot., 30, 1015–1018 (1977).
- 27) Saito E., Daikuhara N., Saito N., Heterocycles, 74, 411–420 (2007).
- 28) Dess D. B., Martin J. C., J. Am. Chem. Soc., 113, 7277–7287 (1991).
- 29) Frincke J. M., Faulkner D. J., J. Am. Chem. Soc., 104, 265–269 (1982); Errata: Idem., Ibid., 104, 5004 (1982).
- 30) He H.-Y., Faulkner D. J., J. Org. Chem., 54, 5822–5824 (1989).
- 31) Fukuyama T., Linton S. D., Tun M. M., Tetrahedron Lett., 31, 5989–5992 (1990).
- 32) Davidson B. S., Tetrahedron Lett., 33, 3721–3724 (1992).
- 33) Parameswaran P. S., Naik C. G., Kanmat S. Y., Pramanik B. N., Indian J. Chem. Sect. B, 37B, 1258–1263 (1998).
- 34) Saito N., Sakai H., Takai E., Muranaka R., Itabashi M., Kubo A., Yazawa K., Mikami Y., Heterocycles, 46, 309–320 (1997).
- 35) Saito N., Sakai H., Suwanborirux K., Pummangura S., Kubo A., Heterocycles, 55, 21–28 (2001).
- 36) Pettit G. R., Knight J. C., Collins J. C., Herald D. L., Pettit R. K., Boyd M. R., Young V. G., J. Nat. Prod., 63, 793–798 (2000).
- 37) Fontana A., Cavaliere P., Wahidulla S., Naik C. G., Cimino G., Tetrahedron, 56, 7305–7308 (2000).
- 38) Suwanborirux K., Personal communication
- 39) Koizumi Y., Kubo A., Suwanborirux K., Saito N., Heterocycles, 57, 2345–2355 (2002).
- 40) Arai T., Takahashi K., Ishiguro K., Yazawa K., J. Antibiot., 33, 951–960 (1980).
- 41) Mikami Y., Yokoyama K., Tageta H., Nakagaki K., Arai T., J. Pharmcobio-Dyn., 4, 282–286 (1981).
- 42) Suwanborirux K., Amnuoypol S., Plubrukan A., Pummangura S., Kubo A., Tanaka C., Saito N., J. Nat. Prod., 66, 1441–1446 (2003).
- 43) Amnuoypol S., Suwanborirux K., Pummangura S., Kubo A., Tanaka C., Saito N., J. Nat. Prod., 67, 1023–1028 (2004).
- 44) Daikuhara N., Tada Y., Yamaki S., Charupant K., Amnuoypol S., Suwanborirux K., Saito N., Tetrahedron Lett., 50, 4276–4278 (2009).
- 45) Saito N., Yoshino M., Charupant K., Suwanborirux K., Heterocycles, 84, 309–314 (2012).
- 46) Debelius H., “Nudibrnchs and Sea Snails Indo-Pacific Field Guide,” 4th ed., Conchbooks, Frankfurt, Germany, 2004, p. 251.
- 47) Charupant K., Suwanborirux K., Amnuoypol S., Saito E., Kubo A., Saito N., Chem. Pharm. Bull., 55, 81–86 (2007).
- 48) “The Asia-Africa Science Platform Program,” Development of effective method for early diagnosis and treatment of intractable disease utilizing characteristics biologically active natural products and enzyme.: ‹https://www.jsps.go.jp/j-aaplat/data/10ichiran_aaplat/saiyokadai_h22/h22/22-9_meijiyakkadai_p_H22houkoku.pdf.›
- 49) Tatsukawa M., Punzalan L. L., Magpantay H. D. S., Villaseňor I. M., Concepcion G. P., Suwanborirux K., Yokoya M., Saito N., Tetrahedron, 68, 7422–7428 (2012).
- 50) Fusetani’s group preliminarily reported a new renieramycin derivative, named renieramycin J,51) but we had previously named the structures of renieramycins J–L (9j–l) in a published paper.42) Thus, structure 9p, named renieramycin J, should be renamed renieramycin P in the Additions and Corrections., J. Nat. Prod., 67, 526 (2004).
- 51) Oku N., Matsunaga S., van Soest R. W. M., Fusetani N., J. Nat. Prod., 66, 1136–1138 (2003).
- 52) He W.-F., Li Y., Feng M.-T., Gavagnin M., Mollo E., Mao S.-C., Guo Y.-W., Fitoterapia, 96, 109–114 (2016).
- 53) Huang R.-Y., Chen W.-T., Kurtàn T., Màndi A., Ding J., Li J., Li X.-W., Guo Y.-W., Future Med. Chem., 8, 17–27 (2016).
- 54) Fukuyama T., Linton S. D., Tun M. M., Tetrahedron Lett., 31, 5989–5992 (1990).
- 55) Fukuyama T., Sachleben R. A., J. Am. Chem. Soc., 104, 4957–4958 (1982).
- 56) Fukuyama T., J. Synth. Org. Chem. Jpn., 46, 801–810 (1988).
- 57) Fukuyama T., Yang L., Ajeck K. L., Sachleben R. A., J. Am. Chem. Soc., 119, 3712–3713 (1990).
- 58) Saito N., Yamauchi R., Kubo A., Heterocycles, 32, 1203–1214 (1990).
- 59) Saito N., Tanaka C., Koizumi Y., Suwanborirux K., Amnuoypol S., Pummangura S., Kubo A., Tetrahedron, 60, 3878–3881 (2004).
- 60) Xu S., Wang G., Zhu J., Shen C., Yang Z., Yu Z., Li Z., Lin T., Sun X., Zhang F., Eur. J. Org. Chem., 2017, 975–983 (2017).
- 61) Lane J. W., Chen Y., Williams R. M., J. Am. Chem. Soc., 127, 12684–12690 (2005).
- 62) Wu Y.-C., Zhu J., Org. Lett., 11, 5558–5561 (2009).
- 63) Liu W., Liao X., Dong W., Yan Z., Wang N., Liu Z., Tetrahedron, 68, 2759–2764 (2012).
- 64) Chen R., Liu H., Chen X., J. Nat. Prod., 76, 1789–1795 (2013).
- 65) Zheng Y., Li X. D., Sheng P.-Z., Yang H.-D., Wei K., Yang Y.-R., Org. Lett., 22, 4489–4493 (2020).
- 66) Chrzanowska M., Grajewska A., Rozwadowska M. D., Chem. Rev., 116, 12369–12465 (2016).
- 67) Magnus P., Matthews K. S., J. Am. Chem. Soc., 127, 12476–12477 (2005).
- 68) Liao X. W., Liu W., Dong W. F., Guan B. H., Chen S. Z., Liu Z. Z., Tetrahedron, 65, 5709–5715 (2009).
- 69) Yokoya M., Fujino K.-S., Saito N., Tetrahedron Lett., 52, 2446–2449 (2011).
- 70) Yokoya M., Fujino K.-S., Yoshida S., Mimura M., Takada H., Saito N., Tetrahedron, 68, 4106–4181 (2012).
- 71) Gonzàlez J. F., Salazar L., de la Cuesta E., Avendaňo C., Tetrahedron, 61, 7447–7455 (2005).
- 72) Yokoya M., Kawachi O., Saito N., Heterocycles, 76, 1497–1509 (2008).
- 73) Chan C., Heid R., Zheng S., Guo J., Zhou B., Furuuchi T., Danishefsky S. J., J. Am. Chem. Soc., 127, 4596–4598 (2005).
- 74) Vincent G., Williams R. M., Angew. Chem. Int. Ed., 50, 8458 (2011).
- 75) Chen X., Zhu J., Angew. Chem. Int. Ed., 46, 3962–3965 (2007).
- 76) Du J., Watanabe K. A., Synth. Commun., 34, 1925–1930 (2004).
- 77) Vincent G., Chen Y., Lane J. W., Williams R. M., Heterocycles, 72, 385–396 (2007).
- 78) Yokoya M., Ito H., Saito N., Chem. Pharm. Bull., 59, 787–792 (2011).
- 79) Yokoya M., Ito H., Saito N., Tetrahedron, 67, 9185–9192 (2011).
- 80) Yokoya M., Kobayashi K., Sato M., Saito N., Mar. Drugs, 13, 4915–4933 (2015).
- 81) Rinehart K. L., Holt T. G., Fregean N. L., Stroh J. G., Keifer P. A., Sun F., Li L., Martin D. G., J. Org. Chem., 55, 4512–4515 (1990).
- 82) Sakai R., Rinehart K. L., Guan Y., Wang A. H.-J., Proc. Natl. Acad. Sci. U.S.A., 89, 11456–11460 (1992).
- 83) Guan Y., Sakai R., Rinehart K. L., Wang A. H.-J., J. Biomol. Struct. Dyn., 10, 793–818 (1993).
- 84) Sakai R., Jares-Erijiman E. A., Manzanares I., Elipe M. V. S., Rinehart K. L., J. Am. Chem. Soc., 118, 9017–9023 (1992).
- 85) Henriquez R., Faircloth G., Cuevas C., “Anticancer Agents from Natural Products,” ed. by Cragg G. M., Kingston D. G., Newman D. J., Taylor & Francis, New York, 2005, pp. 215–223.
- 86) Menchaca R., Martinez V., Rodriguez A., Rodriguez N., Flores M., Gallegp P., Manzanares I., Cuevas C., J. Org. Chem., 68, 8859–8866 (2003).
- 87) Corey E. J., Gin D. Y., Kania R. S., J. Am. Chem. Soc., 118, 9202–9203 (1996).
- 88) Martinez E., Corey E. J., Org. Lett., 2, 993–996 (2000).
- 89) Endo A., Yanagisawa A., Abe M., Tohma S., Kan T., Fukuyama T., J. Am. Chem. Soc., 124, 6652–6654 (2002).
- 90) Kawashi F., Toma T., Inui T., Yokoshima S., Fukuyama T., J. Am. Chem. Soc., 135, 13684–13687 (2013).
- 91) Chen J., Chen X., Bois-Choussy M., Zhu J., J. Am. Chem. Soc., 128, 87–89 (2006).
- 92) He W., Zhang Z., Ma D., Angew. Chem. Int. Ed., 58, 3972–3975 (2019).
- 93) Zheng S., Chan C., Furuuchi T., Wright B. J. D., Zhou B., Guo J., Danishefski S. J., Angew. Chem. Int. Ed., 45, 1754–1759 (2006).
- 94) Fishlock D., Williams R. M., J. Org. Chem., 73, 9594–9600 (2008).
- 95) Ikeda Y., Matsuki H., Ogawa T., Munakata T., J. Antibiot., 36, 1284–1289 (1983).
- 96) Okumoto T., Kawana M., Nakamura I., Ikeda Y., Isagai K., J. Antibiot., 38, 767–771 (1985).
- 97) Cuevas C., Peréz M., Martin M. J., Chicharro J. L., Fernández-Rivas C., Flores M., Francesch A., Gallego P., Zarzuelo M., de la Calle F., Garcia J., Polanco C., Rodriguez I., Manzanares I., Org. Lett., 2, 2545–2548 (2000).
- 98) Cuevas C., Francesch A., Nat. Prod. Pep., 26, 322–337 (2009).
- 99) Herdman W. A., Proc. Trans. Liverpool Biol. Sci., 5, 151–154 (1891), we are grateful to Professor Teruaki Nishikawa of the Nagoya University Museum for his identification of the Thai tunicate.
- 100) Suwanborirux K., Charupant K., Amnuoypol S., Pummangura S., Kubo A., Saito N., J. Nat. Prod., 65, 935–937 (2002).
- 101) Puthongking P., Patarapanichi C., Amnuoypol S., Suwanborirux K., Kubo A., Saito N., Chem. Pharm. Bull., 54, 1010–1016 (2006).
- 102) Sousa S. C. A., Fernandes A. C., Tetrahedron Lett., 50, 6872–6876 (2009).
- 103) Hayer P., Hosten E., Gerter T. L. A., Abrahams A., Bull. Korean Chem. Soc., 30, 1204–1206 (2009).
- 104) Tsujimoto M., Lowtangkitcharoen W., Mori N., Pangkruang W., Puthongking P., Suwanborirux K., Saito N., Chem. Pharm. Bull., 61, 1052–1064 (2013).
- 105) Charupant K., Daikuhara N., Saito E., Amnuoypol S., Suwanborirux K., Owa T., Saito N., Bioorg. Med. Chem., 17, 4548–4558 (2009).
- 106) Sirimangkalakitti N., Chamni S., Charupant K., Chanvorachote P., Mori N., Saito N., Suwanborirux K., J. Nat. Prod., 79, 2089–2093 (2016).
- 107) Martinez E. J., Owa T., Schreiber S. L., Corey E. J., Proc. Natl. Acad. Sci. U.S.A., 96, 3496–3501 (1999).
- 108) Plowright A. T., Schaus S. E., Myers A. G., Chem. Biol., 9, 607–618 (2002).
- 109) Ocio E. M., Maiso P., Chen X., Garayoa M., Stela Álvarez-Fernández S., San-Sequndo L., Vilanova D., López-Corral L., Montero L. C., Hernández-Iglesias T., de Álava E., Galmarini C., Avilés P., Cuevs C., San-Miguel J. F., Blood, 113, 3781–3791 (2009).
- 110) Schlumacher M., Kelkel M., Dicato M., Diederich M., Biotechnol. Adv., 29, 531–547 (2011).
- 111) Toyoshima R., Mori N., Suzuki T., Lowtangkitcharoen W., Suwanborirux K., Saito N., Chem. Pharm. Bull., 64, 966–969 (2016).
- 112) Rath C. M., Janto B., Earl J., Earl J., Ahmed A., Hu F. Z., Hiller L., Dahlgen M., Kreft R., Yu F., Wolff J. J., Kweon H. K., Christiansen M. A., Hakansson K., Williams R. M., Ehrlich G. D., Sherman D. H., Chem. Biol., 6, 1244–1256 (2011).
- 113) Welin E. R., Ngamnithiporn A., Klatte M., Lapointe G., Pototschnig G. M., McDermott M. S. J., Conklin D., Gilmore C. D., Tadross M. S. J., Haley C. K., Negoro K., Gilibstrup E., Grünanger C. U., Allan K. M., Virgil S. C., Slamon D. J., Stoltz B. M., Science, 363, 270–275 (2019).
















