Reviews
Development of Catalytic Reactions for Precise Control of Chemoselectivity
2021 Volume 69 Issue 6 Pages 516-525
Details
2021 Volume 69 Issue 6 Pages 516-525
Catalytic chemoselective reactions of innately less reactive functionalities over more reactive functionalities are described. A cooperative catalyst comprising a soft Lewis acid/hard Brønsted base enabled chemoselective activation of a hydroxyl group over an amino group, allowing for nucleophilic addition to electron-deficient olefins. The reaction could be applicable for a variety of amino alcohols, including pharmaceuticals, without requiring a tedious protection–deprotection process. Chemoselective enolization and subsequent α-functionalization of carboxylic acid derivatives were also achieved by a redox active catalyst through the radical process, providing unnatural α-amino/hydroxy acid derivatives bearing a complex carbon framework and a diverse set of functionalities. The present chemoselective catalysis described herein offers new opportunities to expand the chemical space for innovative drug discovery research.
Chemoselective reactions in which the reactivity of the functional group is precisely controlled by the catalyst are ideal synthetic processes that bypass requirements for tedious protection–deprotection procedures and umpolung.1–4) These types of reactions also comprise a fundamental technology that opens up a new chemical space in terms of affording different products from those produced by conventional methods. Despite the high potential for contributions to both the atom and step economy5,6) of catalytic chemoselective reactions, progress in this area has been limited relative to corresponding stereo- or regioselective reactions. In this review, two types of catalytic chemoselective reactions are described: 1) chemoselective catalytic activation of hydroxyl groups, and 2) chemoselective catalytic α-functionalization of carboxylic acid equivalents and carboxylic acids through the radical processes.
When amino alcohols, which are found in many pharmaceutical substructures, are used as nucleophiles, their reactions with electrophiles generally proceed in an amino group-selective manner owing to the high nucleophilicity of the amino group. Several catalyst-controlled chemoselective reactions of hydroxyl groups over an amino group were recently reported.7–17) The reaction types, however, were limited and remain unexplored. Moreover, the reported reactions include the inevitable formation of stoichiometric amounts of co-products. Thus, we focused on an atom-economical conjugate addition of hydroxyl groups, which is applicable to natural product synthesis.18–20) We envisioned that a soft Lewis acid would activate soft Lewis basic electrophiles, α,β-unsaturated nitriles,21,22) even in the presence of hard Lewis basic hydroxyl and amino groups. Whereas a Lewis acid catalyst would activate electrophiles to facilitate the subsequent coupling of an innately more nucleophilic amino group, a cooperative catalyst comprising soft Lewis acids and hard Brønsted bases would enable simultaneous activation of both soft Lewis basic electrophiles and hard hydroxyl groups, allowing for chemoselective coupling.23,24)
Consistent with this concept, a cooperative catalyst comprising a Cu(I) phosphine complex and lithium base (lithium bis(trimethylsilyl)amide or n-butyllithium) efficiently promoted the conjugate addition of amino alcohol25) (Table 1). Without the cooperative catalyst, an amine-derived conjugate addition proceeded exclusively. The use of strong bases such as lithium bis(trimethylsilyl)amide or n-butyllithium afforded the alcohol-derived product in low yield, suggesting that the combined use of a copper(I) complex and a Brønsted base was essential for obtaining both a high yield and high chemoselectivity. Mesitylcopper26) with a dppe ligand exhibited high catalytic performance, indicating that copper(I) alkoxide is the actual catalytic species. A wide variety of amino alcohols could be used under optimized catalytic conditions. An amino acid-derived β-amino alcohol, L-phenylalaninol, which was not applicable in the previously described reactions,7–16) delivered product 3ad in a high yield with excellent chemoselectivity, and product 3ad was isolated on a gram scale. Relatively acidic aniline N–H did not affect the chemoselectivity (3af, 3ag).27) Congested secondary alcohols also selectively reacted with 1a (3ah). The process could be applied to amino alcohol, possessing a primary alcohol and a highly nucleophilic secondary amine (3ai). Pharmaceuticals, biomolecules, and peptides proved to be good substrates (3aj–3aq).
 |
The high chemoselectivity of the present cooperative catalysis was not confined to hydroxy groups. Chemoselective conjugate addition to soft Lewis basic α,β-unsaturated nitriles over hard Lewis basic tert-butyl acrylate as an additional electrophile was achieved, while a mixture of N-adducts derived from both 1a and 15 were obtained without a catalyst (Chart 1).

An advantage of the chemoselective catalysis is its wide substrate scope. A multistep sequence, including protection of amino groups required for the transformation of the nitrile functionality in some cases and direct catalytic installation of various functional groups onto less nucleophilic hydroxy groups of unprotected amino alcohol, has never been achieved. Thus, we focused on α,β-unsaturated sulfonyl derivatives for direct functional group installation.28–32) α,β-Unsaturated sulfonyl derivatives are often used as labeling reagents because of their highly electrophilic nature, allowing for divergent functional group installation into functionalized molecules. An optimization study based on the previous copper(I) complex/hard Brønsted base catalysis revealed that the silver(I) complex exhibited slightly superior catalytic performance.33)
With the optimized conditions in hand, we next explored the substrate scope. Unlike α,β-unsaturated nitriles, various α,β-unsaturated sulfonyl derivatives bearing functional groups could be used (Table 2). Substituted aryl vinyl sulfones could be used under the optimized conditions and the products were isolated in good yield and could be further transformed through late-stage functionalization (7aa–7ca).34,35) A variety of sulfonamides were applicable, and amino acids, azide, and a fluorescent-labeling dansyl group could be installed onto a hydroxy group in high yield (7ia–7la).
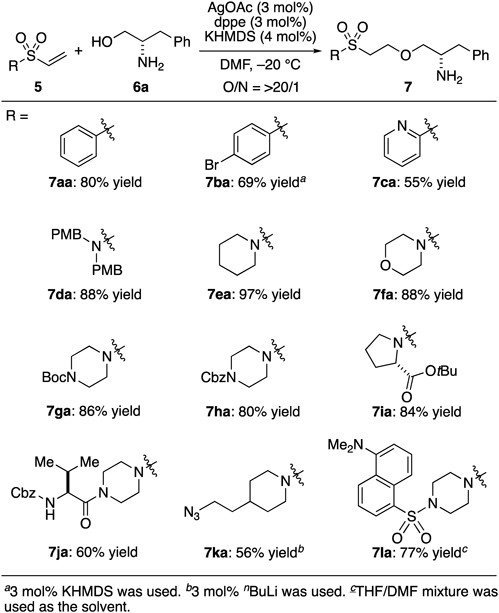 |
Various amino alcohols, including pharmaceuticals and biomolecules, could be used with an extended substrate scope compared with the previous copper catalyzed chemoselective conjugate addition (Table 3). Pharmaceuticals (propranolol, hydroxychloroquine, and ambroxol), amino acid derivatives, and nucleoside derivatives were applicable and hydroxy group adducts 7eq–7ev were isolated chemoselectively. The utility of the present catalytic chemoselective functional group installation method was further enhanced by coupling 5l with ambroxol (6s) (Chart 2). Fluorescent-labeling dansyl group was chemoselectively attached onto hydroxy group over aliphatic and aryl amino groups.
 |

We next envisioned that a hydroxy group at the β-position of the free amino group would be selectively activated through bidentate coordination to a Lewis acidic silver salt, even in the presence of another distinct hydroxy group (Chart 3). After slightly modifying the reaction conditions, almost perfect chemo- and regioselective fluorescent labeling of aminodiol (6w) was achieved using 5l, highlighting the synthetic utility of the free amino group-directed hydroxy group selective reaction (Chart 4).


We further expanded our chemoselective catalytic conjugate addition to α,β-unsaturated carboxylate derivatives.36–39) Although the use of α,β-unsaturated ester as an electrophile for the catalytic conjugate addition of alcohols is more advantageous for further elaboration, examples are extremely limited owing to difficulties in controlling the chemoselectivity between conjugate addition (1,4-addition) and transesterification (1,2-addition). We hypothesized that our cooperative catalyst system comprising a soft Lewis acid/hard Brønsted base would chemoselectively generate a soft metal alkoxide, and thereby soft conjugate addition of alkoxide would be preferred over undesired hard transesterification40) (Chart 5).

High chemoselectivity was observed using amino acid derivatives containing an amide functionality with acidic NH protons and a silver/sodium bis(trimethylsilyl)amide (NaHMDS) catalyst system (Chart 6). Hydroxychloroquine (9b) also delivered O-adduct 10ab in 33% yield with high chemoselectivity (Chart 6).

Carbonyl compounds are ubiquitous in organic compounds, and their transformation reactions have been classically studied. In particular, the activation of carbonyl compounds by deprotonation of the α-proton (enolization) followed by reaction with an electrophile plays a central role in synthetic organic chemistry as a powerful carbon–carbon bond-forming reaction. In recent years, many stereoselective reactions using metal catalysts and organocatalysts offering easy access to chiral molecules have been reported.41–45) Among carbonyl compounds, carboxylic acids and their derivatives, such as esters and amides, are abundant in nature and one of the most important functional groups found in many pharmaceuticals and functional molecules, including amino acids and fatty acids. They also allow for divergent transformation compared with corresponding ketones and other carbonyls. In recent years, carboxylic acids have been used as an alkyl radical precursor via a decarboxylation reaction.46–48) The intrinsic low acidity of the α-protons of carboxylic acids and their derivatives, however, makes catalytic activation challenging. Enolization of carboxylic acid conventionally relies on the use of more than 2 equivalents of a strong Brønsted base due to the innate Brønsted acidic carboxylic acid functionality. Remarkable advances have been made by the combined use of a Lewis acid and weak Brønsted base, so-called soft enolization. A stoichiometric or catalytic amount of boron reagent efficiently generated 1,1-enediolate with a stoichiometric amount of Brønsted base, followed by addition to polar electrophiles, such as aldehydes and imines.49–52)
3.1. Chemoselective Catalytic α-Amination of Acylpyrazolesα-Amino acids are widely used in the field of synthetic organic chemistry as functional molecules and building blocks for pharmaceuticals and other applications.53) Middle molecular peptide drugs have recently attracted much attention and the development of efficient synthetic methods for unnatural α-amino acids is in high demand.54–59) In particular, α-amination of carboxylic acid derivatives60–62) is anticipated as a new method for synthesizing unnatural α-amino acids to complement conventional methods, as exemplified by Strecker-type reactions63–65) and alkylation of glycine derivatives.66–68) We focused on acylpyrazoles as carboxylic acid equivalents.69,70) The α-proton of acylpyrazole exhibits high acidity due to the weak amide conjugation and forms a chelate structure with a proper Lewis acid between the nitrogen atom on the pyrazole ring and the oxygen atom of the carbonyl group, allowing for chemoselective enolization over an innately more acidic functionality such as ketone. Acylpyrazoles can be easily synthesized from carboxylic acids under mild conditions and converted to various oxidation states. We anticipated that a proper Lewis acidic transition metal catalyst that can generate metal nitrenoide species from iminoiodinane would deliver the α-amination product.71) Extensive screening of the conditions revealed that cationic copper(II) triflate was a competent catalyst with a broad substrate scope72) (Table 4). A variety of functional groups (alkene, alkyne, alkyl halides, oxygen functional groups, nitrogen functional groups, boronic acid esters) were allowed under the optimized conditions and α-amino acid derivatives with a diverse set of carbon skeletons and functional groups in the side chains were synthesized. Our α-amination was applicable to late-stage functionalization and lithocholic acid derivatives bearing complex carbon skeletons, delivering α-amino acid derivatives in moderate yields (13r).
 |
When a substrate having a nitroalkyl functionality was treated with a catalytic amount of triethylamine in D2O, deuteration proceeded selectively at the α-position of the nitro functionality (Chart 7). On the other hand, the amination proceeded chemoselectively at the α-position of acylpyrazole under the optimized reaction conditions (Chart 7). α-Amination of acylpyrazoles also proceeded even in the presence of other electron-withdrawing functionalities, ketones, esters, nitriles, sulfones, and phosphates73) (Table 5).

 |
Acylpyrazoles were applied to the α-oxidation reaction. As an oxygenating agent, a commercially available 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) free radical was selected for the aerobic α-oxidation reaction.74–81) Although cationic copper(II) triflate, which was the optimal catalyst for the α-amination reaction, exhibited low catalytic performance, copper(I) chloride under an oxygen atmosphere afforded the α-oxidation product in high yield.82) A series of control experiments revealed that an in situ-generated copper(II) peroxo complex under an oxygen atmosphere would be an actual catalytic species and serve as a Lewis acid/Brønsted base cooperative catalyst83) (Chart 8).

We then investigated late-stage α-oxidation of a variety of pharmaceutical agents and natural products (Table 6). The nonsteroidal anti-inflammatory drugs felbinac, isoxepac, diclofenac, indomethacin, and zomepirac derivatives, were efficiently oxidized under mild conditions (16a–16e). An α-lipoic acid derivative, having an oxidation-labile cyclic disulfide functionality, was chemoselectively oxidized at the α-position over an oxidation-labile cyclic disulfide functionality (16f). α-Aliphatic pharmaceutical agents and natural product derivatives were also applicable to the present catalysis and the corresponding α-hydroxy acid derivatives were isolated in high to moderate yield (16g–16j)
 |
In previous reactions, acylpyrazoles were used as carboxylic acid equivalents for efficient chemoselective enolate formation. Although these reactions exhibited a wide range of substrate generality, ubiquitous carboxylic acids should ideally be used without any preactivation. Conventional methods, however, require stoichiometric amounts of external base to neutralize the Brønsted acidity characteristic of carboxylic acids. In addition, a catalytic system applicable to radical reactions that can complement the classical ionic type reaction has not been achieved. Optimization studies revealed that molecular sieves significantly affected the reaction rate and the combined use of iron(II) complex and 4 Å molecular sieves efficiently catalyzed the α-oxidation of carboxylic acid.84) The present catalysis was hardly affected by a change in the electron density in the aromatic ring at the α-position of carboxylic acid (Table 7). Increasing the amount of catalyst and TEMPO afforded the desired product 18ca in high yield when a bulky substrate was used. Notably, various functional groups, including iodide, thioether, Boc-protected aniline, hydroxy groups, and benzyl bromide, could be used. Several pharmaceuticals and functionalized TEMPO derivatives were applicable to the present catalysis. β,γ-Unsaturated carboxylic acid afforded the γ-oxidized product 18ζa in high yield.
 |
The chemoselectivity of the present catalysis was confirmed by adding carbonyl compounds such as ketones, esters, and amides under optimal conditions (Chart 9). As a result, the chemoselective oxidation of carboxylic acids was achieved and product 18aa was isolated in high yield with the recovery of carbonyl compounds unchanged.

We next focused on the detailed reaction mechanism. Initial-rate kinetic studies of the reaction of 17a and TEMPO were performed (Fig. 1a). The negative order kinetic dependency with respect to carboxylic acid 17 would be due to the acidic proton of carboxylic acid inhibiting the enolization step. The zeroth dependency on TEMPO indicated that TEMPO was not involved in limiting the turnover. The reaction rate displayed 0.7th dependency on the catalyst, suggesting that an inactive oligomeric species would exist in equilibrium with an active monomeric species. When two parallel reactions were performed under optimized conditions using 1a and 1a(d2), a relatively large kinetic isotope effect (kH/kD = 2.01) was observed, indicating that enolization of carboxylic acid makes a major contribution to limiting the turnover (Fig. 1b).

A series of mechanistic studies led to the proposed catalytic cycle shown in Fig. 1c. Initially, iron(II) species would be oxidized by TEMPO to form the iron(III) species. Subsequently, the iron carboxylate species III would be generated through an exchange of counter anions. The iron carboxylate species III is in equilibrium with the inactive oligomer IV, and the ligand would promote regeneration of the active species. Enolization would be achieved with the assistance of alkali metal carboxylate generated from MS4A, generating the redox active heterometallic enediolate V. The radical species VI generated by resonance with this enediolate would couple with TEMPO to produce the product with generation of the iron(II) species.
We also confirmed that even under the condition of using 20 mol% sodium acetate instead of MS4A, the target product was obtained in high yield (Chart 10), indicated that the functionalization of carboxylic acids was achieved without the need for neutralization of Brønsted acidity by stoichiometric amounts of base.

In conclusion, we developed two types of catalyst-controlled chemoselective reactions using a cooperative catalyst comprising a soft Lewis acid/hard Brønsted base and a redox active transition metal catalyst for a radical process. The innately less reactive hydroxy group was activated through deprotonation in the presence of the amino group, generating soft metal-conjugated hard alkoxide species. Acylpyrazoles and carboxylic acid could be chemoselectively activated by a redox active copper and iron/alkali metal catalyst without external addition of stoichiometric amount of Brønsted bases, allowing for chemoselective α-functionalization through the radical process. The present chemoselective radical process expanded to several carbon–carbon bond-forming reactions.85–88) The present chemoselective catalysis is highly promising for the discovery and design of new drugs.
I would like to express my sincere gratitude to Prof. Takashi Ohshima (Kyushu University) for his guidance and fruitful suggestions, and for providing a relaxed research environment. I am deeply grateful to all my coworkers whose names appear in the reference section for their critical contribution to the present achievement. This work was supported by Grants-in-Aid for Research Activity Start-up, Young Scientists (B), Scientific Research (C), Challenging Research (Exploratory), and Scientific Research on Innovative Areas from the Japan Society for the Promotion of Science.
The authors declare no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2020 Pharmaceutical Society of Japan Award for Young Scientists.