Abstract
Sulfur K-edge X-ray absorption near-edge structure (XANES) spectroscopy was evaluated for its ability to detect non-conventional C–H▪▪▪S hydrogen bonds in crystals of the sulfur-containing penam antibiotics ampicillin and amoxicillin. The XANES spectra of the nearly isomorphous crystals of ampicillin trihydrate and amoxicillin trihydrate were very similar, whereas that of ampicillin anhydrate displayed unique features. Single-crystal X-ray structure analyses revealed that the C–H▪▪▪S hydrogen bond geometries and the chemical types of the hydrogen donors differed between the isomorphous trihydrate crystals and ampicillin anhydrate crystal. These observations demonstrate that the shapes of the sulfur K-edge XANES spectra are dependent on the nature of the C–H▪▪▪S hydrogen bonds. Sulfur K-edge XANES spectroscopy shows promise for use in the detection and analysis of non-covalent interactions, including hydrogen bonds to sulfur atoms, within active pharmaceutical ingredients.
Introduction
Non-covalent bonding plays a critical role in molecular recognition. Not only is it involved in the binding of active pharmaceutical ingredient (API) molecules to their receptors but also in the formation of API crystals, amorphous forms and solid dispersions. The most notable non-covalent bonding interaction is the hydrogen bond that is formed between donor atoms with high electronegativity and acceptor atoms with lone pairs, such as oxygen and nitrogen. In addition to these conventional strong hydrogen bonds, non-conventional weak hydrogen bonds, such as C–H▪▪▪O,1) C–H▪▪▪N,2) C–H▪▪▪π3) and C–H▪▪▪S,4) have also been recognized as important specific intermolecular interactions.5,6)
Non-conventional weak hydrogen bonds have often been investigated using crystal structure analysis. This is because single-crystal X-ray analysis reveals atomic coordinates with high precision. Recently, X-ray absorption near-edge structure (XANES) spectroscopy7,8) has been applied to the analysis of non-covalent conventional hydrogen bonds and halogen bonds of APIs.9,10) In X-ray absorption spectroscopy, X-ray absorbances increase sharply when the X-ray energy is high enough to excite electrons in the core orbitals, such as the K-shells. These sharp increases in absorption, visible in the spectra, are called absorption edges. The shapes of the higher energy regions above these edges, XANES, are sensitive to the interactions of the X-ray absorbing atom. This sensitivity is a result of the interactions affecting the atom’s electronic state, because the interactions are accompanied by the overlaps of electron orbitals and the overlaps would change the energies and shapes of the electron orbitals of X-ray absorbing atoms. XANES spectroscopy is the method with a high element specificity and is suitable for detecting specifically the interatomic interactions at the X-ray absorbing atoms. The typical target elements of XANES spectroscopy in pharmaceutical research are chlorine,9–11) bromine12,13) and phosphorus,14) although XANES may be applied, in principle, to all elements.
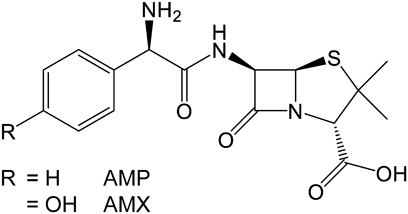
In this study, we evaluate the ability of sulfur K-edge XANES spectroscopy to detect the formation of non-conventional C–H▪▪▪S hydrogen bonds in API crystals, together with single-crystal X-ray structure analysis. Crystals of the sulfur-containing penam antibiotics ampicillin (AMP) and amoxicillin (AMX) were used as model API crystals (Fig. 1).
Results and Discussion
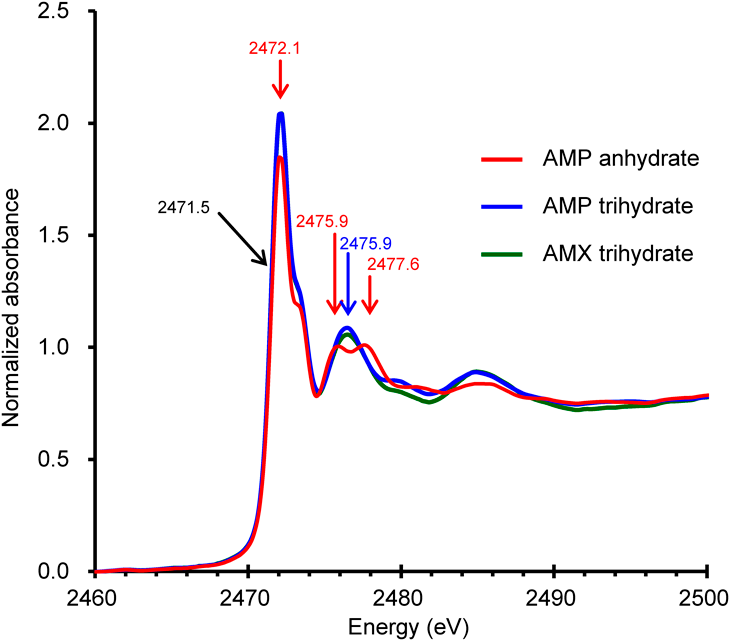
The sulfur K-edge XANES spectra of AMP anhydrate, AMP trihydrate and AMX trihydrate all have absorption edge energies (where the second derivatives of the sharp rises are zero) of 2471.5 eV (Fig. 2). The spectra of AMP trihydrate and AMX trihydrate are nearly identical; the only exception is that the absorbances around 2480 eV are slightly lower for AMX trihydrate than for AMP trihydrate. Although the chemical structures of AMP and AMX differ by a hydroxy group at the phenyl moiety, the hydroxyl group is far from the sulfur atom and thus may not affect its electronic state. The AMX trihydrate crystal and the AMP trihydrate crystal are almost isomorphous (Table 1). This indicates that the sulfur atoms in each crystal share a common electronic state, explaining why the spectra of AMP trihydrate and AMX trihydrate are remarkably similar. In contrast, the spectrum of AMP anhydrate has unique features. The height of the largest peak of AMP anhydrate (at 2472.1 eV) is lower by approximately 10% than that of AMP trihydrate. In addition, two clearly separated peaks were observed at 2475.9 and 2477.6 eV for AMP anhydrate, whereas only a single peak was observed (at 2476.5 eV) for AMP trihydrate. These spectral differences are thought to be caused by differences in the molecular conformations or interatomic interactions of the X-ray absorbing sulfur atom.
Table 1. Statistics of Single-Crystal X-Ray Analyses
| AMP anhydrate | AMP trihydrate | AMX trihydrate |
|---|
| Chemical formula | C16H19N3O4S | C16H19N3O4S·3(H2O) | C16H19N3O5S·3(H2O) |
| Temperature (°C) | 20 | −180 | 20 | −173 | 20 | −180 |
| Space group | P 21 | P 21 | P 212121 | P 212121 | P 212121 | P 212121 |
| Cell parameter (Å, degree) | | | | | | |
| a | 11.9934(3) | 11.9228(2) | 6.6700(2) | 6.63522(19) | 6.68500(10) | 6.65420(10) |
| b | 6.19990(10) | 6.09797(10) | 15.5222(5) | 15.3652(4) | 15.7628(2) | 15.6427(2) |
| c | 12.4009(3) | 12.4145(2) | 18.9329(6) | 18.9107(5) | 18.8278(2) | 18.7669(2) |
| α | 90 | 90 | 90 | 90 | 90 | 90 |
| β | 114.688(3) | 114.379(2) | 90 | 90 | 90 | 90 |
| γ | 90 | 90 | 90 | 90 | 90 | 90 |
| Cell Volume (Å3) | 837.82(4) | 822.12(3) | 1960.18(11) | 1927.97(9) | 1983.97(4) | 1953.44(4) |
| Z/Z′ | 2/1 | 2/1 | 4/1 | 4/1 | 4/1 | 4/1 |
| Data collection | | | | | | |
| Crystal size (mm) | 0.230 × 0.040 × 0.050 | 0.300 × 0.050 × 0.020 | 0.250 × 0.015 × 0.015 | 0.250 × 0.015 × 0.015 | 0.501 × 0.114 × 0.212 | 0.531 × 0.112 × 0.050 |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| (sinθ/λ) max (Å−1) | 0.698 | 0.714 | 0.714 | 0.733 | 0.696 | 0.696 |
| No. of reflections | | | | | | |
| Measured | 53644 | 37501 | 35646 | 45663 | 99024 | 98272 |
| Unique: all | 4557 | 4698 | 5360 | 5938 | 5387 | 5284 |
| F > 4σ(F) | 3967 | 4499 | 4251 | 5364 | 5025 | 5154 |
| Rint | 0.0393 | 0.0298 | 0.0458 | 0.0438 | 0.03577 | 0.0277 |
| Refinement | | | | | | |
| R (F) [F > 4σ(F)] | 0.0329 | 0.0257 | 0.0347 | 0.0332 | 0.0286 | 0.0242 |
| wR (F2) (all) | 0.1020 | 0.0810 | 0.0811 | 0.0785 | 0.0915 | 0.0831 |
| Goodness-of-fit | 0.793 | 0.727 | 0.923 | 1.060 | 0.801 | 0.793 |
| Δρmax, Δρmin | 0.22, −0.15 | 0.35, −0.16 | 0.18, −0.17 | 0.42, −0.22 | 0.26, −0.15 | 0.35, −0.16 |
| CCDC deposition number | 2172196 | 2172195 | 2172197 | 2172198 | 2172200 | 2172199 |
The superposition of the molecular structures of AMP and AMX, fixed at the penam moieties, shows that the conformations of the aromatic moieties differ by 0.4–1.5 Å (Fig. 3). The aromatic moieties and the sulfur atoms are separated by six covalent bonds and an intramolecular distance of approximately 5–6 Å, which is much larger than the sum of their van der Waals radii. This indicates that the differences in conformation would have a negligible effect on the electronic states of the sulfur atoms and would not lead to differences in the XANES spectra. Therefore, non-covalent intermolecular interactions of the sulfur atoms are presumed to cause the differences in the XANES spectra.
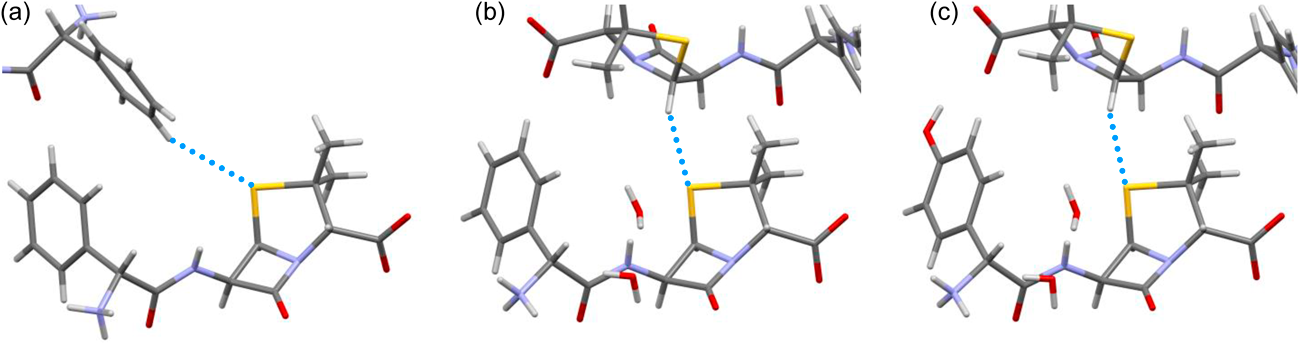
The sulfur atoms of these antibiotics form close interatomic contacts with the hydrogen atoms of neighboring molecules in the crystal (Fig. 4, Table 2). In the AMP anhydrate crystal, the hydrogen atom of the benzene ring is located 3.009 Å from the sulfur atom (at 20 °C). In the AMP trihydrate and AMX trihydrate crystals, the hydrogen atoms of the β-lactam moieties are located 2.915 and 3.069 Å from the sulfur atoms, respectively. All of these H▪▪▪S distances are close to, or smaller than, the sum of van der Waals radii of sulfur (1.8 Å) and hydrogen (1.2 Å). The distances are slightly shorter in crystals at −180 or −173 °C where atomic coordinates were refined more precisely because of the reduced thermal vibrations of atoms. The angle of the C–H▪▪▪S contact in the AMP anhydrate crystal is 141.1° at 20 °C, and those of the AMP trihydrate and AMX trihydrate crystals are slightly larger by approximately 10°. The distances and angles of the C–H▪▪▪S contacts in both the AMP and AMX crystals fell within the previously reported ranges of 2.9–3.3 Å and 126–180°, respectively, which were determined based on a survey of published crystal structures.15) When the atomic coordinates of the hydrogen atoms participating in the C–H▪▪▪S contacts were freely refined, the geometries of the contacts remained within the reported ranges (Supplementary Table S1). This indicates that the contacts can be regarded as the C–H▪▪▪S hydrogen bonds.
Table 2. Geometries of C–H▪▪▪S Hydrogen Bonds
| Crystal | AMP anhydrate | AMP trihydrate | AMX trihydrate |
|---|
| Temperature (°C) | 20 | −180 | 20 | −173 | 20 | −180 |
| C–H distance (Å)* | 0.930 | 0.950 | 0.980 | 1.000 | 0.980 | 1.000 |
| H▪▪▪S distance (Å) | 3.009 | 2.968 | 2.915 | 2.834 | 3.069 | 2.997 |
| C–H▪▪▪S angle (degree) | 141.1 | 139.2 | 152.6 | 153.0 | 155.1 | 154.9 |
* C–H distances in the C–H▪▪▪S hydrogen bonds were fixed to these values during crystallographic refinement using SHELXL.
The formation of C–H▪▪▪S hydrogen bonds would be expected to affect the shapes and energies of sulfur’s electron orbitals, which would change the appearance of the sulfur K-edge XANES spectra. The observed differences in the XANES spectra may be caused by differences in geometries of the C–H▪▪▪S hydrogen bonds and by the types of hydrogen donors (either aliphatic or aromatic carbon atoms). Although the precise relationship between the geometries of hydrogen bonds and the XANES shapes remained to be elucidated, XANES spectroscopy is therefore a sensitive method for specifically investigating the formation of the non-covalent weak bonding of X-ray absorbing atoms in APIs. This characteristic of XANES spectroscopy promises to be highly useful for evaluating the molecular interactions of APIs containing specific elements in pharmaceutical formulations, such as solid dispersions and co-amorphous forms, in which large amounts of various excipients may coexist with the APIs.
Conclusion
XANES spectroscopy and single-crystal X-ray analyses revealed that the shapes of the sulfur K-edge XANES spectra of the penam antibiotics AMP and AMX differed depending on the nature of the non-conventional C–H▪▪▪S hydrogen bonds within the crystals. Because XANES spectroscopy is a highly element-specific method that can analyze not only crystals but also amorphous solids and solutions, it would be widely applicable to the detection and analysis of non-covalent intermolecular interactions of APIs containing specific elements in pharmaceutical formulations.
Experimental
MaterialsAMP anhydrate (purity >96%) was purchased from Merck (Darmstadt, Germany). AMP trihydrate and AMX trihydrate (both purities >98%) were purchased from Tokyo Chemical Industry Col, Ltd. (Tokyo, Japan). All other reagents used were of the highest grade commercially available.
Preparations of AMP and AMX Single CrystalsSingle crystals of AMP anhydrate were picked up from the purchased powders. Single crystals of AMP trihydrate were prepared by recrystallization, as follows. Crystal powders of AMP trihydrate (250 mg) were suspended in 5 mL of water. Aqueous ammonia (37 µL of a 30% (w/w) solution) was added and the mixture was agitated with a vortex mixer for 5 min. The pH of the resulting suspension was 8.1. The suspension was filtered using a disc filter with a 0.45-µm pore size, and the filtrate was slowly evaporated at room temperature. Needle-shaped crystals of AMP trihydrate were obtained within a few days.
Single crystals of AMX trihydrate were prepared by recrystallization, as follows. Amoxicillin trihydrate (300 mg) was dissolved in aqueous hydrochloric acid (30 mL of a 1.0 M solution). The resulting mixture was adjusted to pH 4.7 with 5.0 M aqueous sodium hydroxide and then filtered using a disc filter with a 0.45-µm pore size. The filtrate was left unsealed at room temperature. Plate-shaped crystals appeared within 1 d.
Sulfur K-Edge XANES SpectroscopyXANES measurements were performed at the BL5S1 beamline of the Aichi Synchrotron Radiation Center (Aichi, Japan). Crystallinities of AMP anhydrate, AMP trihydrate and AMX trihydrate powders were 96, 95 and 93% as estimated from powder X-ray diffraction (PXRD) profiles (Supplementary Fig. S1). The crystal powders, approximately 5 mg, were put on conductive and adhesive carbon tapes (JEOL Ltd., Tokyo, Japan; thickness 0.16 mm) and were set in the X-ray absorption spectroscopy chamber. The air in the chamber was substituted with helium. The X-ray energy was calibrated with the XANES spectra of potassium sulfate crystals. XANES spectra were recorded at 25 °C in the total electron yield mode. The XANES measurements were completed within 150 min. The dehydrations of the AMP trihydrate and AMX trihydrate during this time period were determined by the thermal gravimetry to be less than 0.05 water molecule per AMP or AMX molecule (Supplementary Fig. S2), confirming that the dehydrations had little effect on the measurement results. Radiation damages were presumed to be negligible, because the coloring of the samples after the measurements were unrecognizable by the visual inspection and because the damages of the APIs at this synchrotron facility were shown to be little by the facts that the repeated measurements on same samples gave same XANES spectra.10,13,14) ATHENA16) was used to process the XANES spectra; the background was subtracted and the spectra were normalized so that the sharp increase at the edge was 1.
Single-Crystal X-Ray Structure AnalysesX-ray diffraction data of single crystals were collected using an XtaLAB P200 system diffractometer (Rigaku Co., Ltd., Tokyo, Japan) with Mo Kα X-rays at 20 °C and also cryo-temperatures of −173 or −180 °C to obtain crystal structures with highly precise coordinates. Structure determinations and crystallographic refinements were performed using SHELXT,17) Oscail,18) SHELXL19) and shelXle.20) All hydrogen atoms were well-defined in the difference Fourier maps. Non hydrogen atoms were refined freely with anisotropic atomic displacement parameters and hydrogen atoms were refined as riding on their parent atoms. The crystal forms of AMP anhydrate and AMP trihydrate were the same as those reported before,21,22) whereas the crystal structure of AMX trihydrate is reported for the first time in this manuscript. The crystal structures and diffraction data were deposited in the Cambridge Crystallographic Data Centre (CCDC) database. Statistics of the single-crystal X-ray structure analyses and CCDC numbers are summarized in Table 1.
Acknowledgments
The synchrotron radiation experiments were conducted at the BL5S1 beam line of the Aichi Synchrotron Radiation Center, Aichi Science & Technology Foundation (approval numbers 201905036 and 2021L3001). This work was supported by the Japan Society for the Promotion of Science KAKENHI (Grant Nos. 20K07001 and 20K16058).
Conflict of Interest
The authors declare no conflicts of interest.
Supplementary Materials
This article contains supplementary materials.
References
- 1) Scheiner S., Struct. Chem., 30, 1119–1128 (2019).
- 2) Bosch E., Bowling N. P., Oburn S. M., Acta Crystallogr. C, 77, 485–489 (2021).
- 3) Nishio M., J. Mol. Struct., 1018, 2–7 (2012).
- 4) Fargher H. A., Sherbow T. J., Haley M. M., Johnson D. W., Pluth M. D., Chem. Soc. Rev., 51, 1454–1469 (2022).
- 5) Watanabe C., Okiyama Y., Tanaka S., Fukuzawa K., Honma T., Chem. Sci., 12, 4722–4739 (2021).
- 6) Bala S. C., Haque N., Pillalamarri V., Reddi R., Kashyap R., Marapaka A. K., Addlagatta A., Int. J. Biol. Macromol., 129, 523–529 (2019).
- 7) Henderson G. S., de Groot F. M. F., Moulton B. J. A., Rev. Mineral. Geochem., 78, 75–138 (2014).
- 8) Arisawa M., Chem. Pharm. Bull., 67, 733–771 (2019).
- 9) Ito M., Suzuki H., Noguchi S., Cryst. Growth Des., 20, 4892–4897 (2020).
- 10) Suzuki H., Iwata M., Ito M., Noguchi S., Mol. Pharm., 18, 3475–3483 (2021).
- 11) Ito M., Shiba R., Suzuki H., Noguchi S., J. Pharm. Sci., 109, 2095–2099 (2020).
- 12) Suzuki H., Tomita A., Ito M., Noguchi S., Chem. Pharm. Bull., 70, 182–186 (2022).
- 13) Huang Z., Suzuki H., Ito M., Noguchi S., Int. J. Pharm., 625, 122057 (2022).
- 14) Ito N., Hashizuka T., Ito M., Suzuki H., Noguchi S., Pharm. Res., 38, 2147–2155 (2021).
- 15) Fargher H. A., Sherbow T. J., Haley M. M., Johnson D. W., Pluth M. D., Chem. Soc. Rev., 51, 1454–1469 (2022).
- 16) Ravel B., Newville M., J. Synchrotron Radiat., 12, 537–541 (2005).
- 17) Sheldrick G. M., Acta Crystallogr. A, 71, 3–8 (2015).
- 18) McArdle P., J. Appl. Cryst., 50, 320–326 (2017).
- 19) Sheldrick G. M., Acta Crystallogr. C, 71, 3–8 (2015).
- 20) Hübschle C. B., Sheldrick G. M., Dittrich B., J. Appl. Cryst., 44, 1281–1284 (2011).
- 21) Boles M. O., Girven R. J., Acta Crystallogr. B, 32, 2279–2284 (1976).
- 22) Burley J. C., van de Streek J., Stephens P. W., Acta Crystallogr. Sect. E Struct. Rep. Online, 62, o797–o799 (2006).