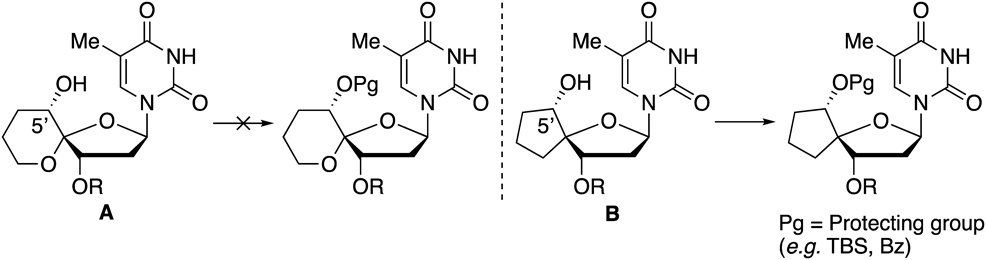
Experimental
GeneralAll the moisture-sensitive reactions were conducted in well-dried glassware in an N2 atmosphere. The IR spectra were recorded on a JASCO FT/IR-4200 spectrometer. The 1H-, 13C-, and 31P-NMR spectra were recorded on JEOL JNM-AL300, JNM-ECS400, and JNM-ECA500 spectrometers. The chemical shifts were reported in part per million (ppm), relative to internal tetramethylsilane (δ = 0.00 ppm), residual CHCl3 (δ = 7.26 ppm), CHD2OD (δ = 3.31 ppm), or CHD2CN (δ = 1.94 ppm) for 1H-NMR. For 13C-NMR, the chemical shifts were reported in ppm, relative to chloroform-d1 (δ = 77.0 ppm), methanol-d4 (δ = 49.0 ppm), or acetonitrile (MeCN)-d3 (δ = 1.32 ppm). For 31P-NMR, the chemical shifts were reported in ppm, relative to 5% H3PO4 (δ = 0.0 ppm) as an external standard. The oligonucleotides were synthesized on a 0.2- and 1.0-µmol scale with an automated DNA synthesizer (Gene Design nS-8). The UV melting experiments were performed on SHIMADZU UV-1650PC and UV-1800 spectrometers that were equipped with Tm analysis accessory quartz cuvettes of 1-cm optional path lengths. The matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) mass spectra of all the new compounds were recorded on a JEOL SpiralTOF JMS-S3000 instrument. MALDI-TOF mass spectra of all the new oligonucleotides were recorded on a Bruker Daltonics Autoflex speed TOF mass spectrometer. For the column chromatography, silica gel PSQ-100B columns (Fuji Silysia Chemical Ltd., Aichi, Japan) were utilized. The progress of the reaction was monitored via analytical TLC on pre-coated glass sheets (Silica gel 60 F254 by Merck, Germany). For the HPLC, SHIMADZU CBM-20 A, DGU-20A3, LC-20AT, CTO-20 A, SPD-20 A, and FRC-10A were utilized. A SHIMADZU UV-1800 spectrometer was utilized for the UV absorbance measurement.
Syntheses of Phosphoramidite (9)1-[(5R)-3-O-tert-Butyldimethylsilyl-4,5-C-diallyl-β-D-2-deoxyribofuranosyl]thymine (3)IBX (2.66 g, 9.51 mmol) was added to a solution containing 229) (1.26 g, 3.17 mmol) in dry MeCN (30 mL) under N2 atmosphere, at room temperature. The reaction mixture was refluxed for 1.5 h, after which the suspension was cooled to room temperature and filtered with Celite®. The filtrate was concentrated in vacuo, and the obtained residue (1.34 g) was dissolved in dry CH2Cl2 (35 mL) in the N2 atmosphere. Next, allyltrimethylsilane (1.6 mL, 10 mmol) and BF3·OEt2 (0.64 mL, 5.10 mmol) were added to this solution at 0 °C, and the reaction mixture was stirred for 3 h at room temperature. After quenching with sat. NaHCO3 aq. at 0 °C, the whole mixture was diluted with CHCl3. Thereafter, the solution was washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The residue (1.22 g) was purified via flash column chromatography (n-hexane–ethyl acetate (AcOEt) = 3 : 2) to yield 3 (865 mg, 63%, two steps from 2) as a white foam.
1H-NMR (CDCl3) δ: 0.11 (3H, s), 0.11 (3H, s), 0.92 (9H, s), 1.92 (3H, d, J = 1.0 Hz), 2.23–2.30 (2H, m), 2.34 (1H, dd, J = 14.5, 8.0 Hz), 2.42 (1H, dt, J = 13.5, 7.0 Hz), 2.50 (1H, dd, J = 13.0, 5.0 Hz), 2.60 (1H, dd, J = 14.0, 6.5 Hz), 3.63 (1H, ddd, J = 10.0, 4.0, 2.5 Hz), 4.64 (1H, dd, J = 7.0, 5.0 Hz), 5.02–5.30 (4H, m), 5.81–5.94 (2H, m), 6.13 (1H, t, J = 6.5 Hz), 7.54 (1H, d, J = 1.0 Hz), 8.00 (1H, s); 13C-NMR (CDCl3) δ: −5.0 (s), −4.2 (s), 12.7 (s), 18.0 (s), 25.8 (s), 36.1 (s), 36.4 (s), 41.2 (s), 73.3 (s), 73.5 (s), 85.4 (s), 90.7 (s), 110.8 (s), 118.4 (s), 118.8 (s), 134.2 (s), 135.2 (s), 137.2 (s), 150.5 (s), 164.2 (s); IR (KBr) cm−1: 3460, 3200, 3070, 2960, 2930, 2900, 2860, 1690, 1470, 1440, 1410, 1360, 1280, 1260, 1210; HRMS (MALDI) m/z 459.2286 (Calcd for C22H36N2NaO5Si: 459.2291).
1-[(2R,4S,5R,10S)-4-tert-Butyldimethylsilyloxy-10-hydroxy-1-oxaspiro[4.5]dec-7-en-2-yl]thymine (4)Grubbs second generation catalyst (12 mg, 0.015 mmol) was added to a solution 3 (1.31 g, 3.00 mmol) in dry CH2Cl2 (30 mL) under N2 atmosphere at room temperature, and the reaction mixture was refluxed for 4 h. The reaction mixture was cooled to room temperature and filtered with Celite®. The filtrate was concentrated in vacuo, and the residue (1.43 g) was purified via flash column chromatography (n-hexane–AcOEt = 3 : 2) to yield 4 (1.15 g, 94%) as a white foam.
1H-NMR (methanol-d4) δ: 0.08 (3H, s), 0.09 (3H, s), 0.90 (9H, s), 1.84 (3H, d, J = 1.0 Hz), 2.15 (1H, ddd, J = 13.5, 6.0, 3.0 Hz), 2.18–2.32 (4H, m), 2.38 (1H, ddd, J = 13.5, 8.0, 5.5 Hz), 3.77 (1H, dd, J = 9.5, 6.0 Hz), 4.55 (1H, dd, J = 5.5, 3.0 Hz), 5.54 (2H, s), 6.24 (1H, dd, J = 8.0, 6.0 Hz), 8.06 (1H, d, J = 1.0 Hz); 13C-NMR (methanol-d4) δ: −5.0 (s), −4.6 (s), 12.6 (s), 18.9 (s), 26.3 (s), 33.2 (s), 33.5 (s), 42.2 (s), 70.4 (s), 75.1 (s), 85.2 (s), 89.3 (s), 111.5 (s), 125.3 (s), 125.3 (s), 138.6 (s), 152.4 (s), 166.4 (s); IR (KBr) cm−1: 3390, 3180, 3030, 2950, 2930, 2900, 2860, 1680, 1470, 1430, 1360, 1330, 1280, 1260, 1210; HRMS (MALDI) m/z 431.1973 (Calcd for C20H32N2NaO5Si: 431.1978).
1-[(2R,4S,5R,6S)-4-tert-Butyldimethylsilyloxy-6-hydroxy-1-oxaspiro[4.5]decan-2-yl]thymine (5)10% Pd/C (268 mg) was added to 4 (1.15 g, 2.81 mmol) in tetrahydrofuran (THF) (30 mL). After stirring the reaction mixture under H2 atmosphere for 1 h at room temperature, the mixture was filtered with Celite®, after which the filtrate was concentrated in vacuo. The residue (1.14 g) was purified via flash column chromatography (n-hexane–AcOEt = 1 : 1) to yield 5 (1.00 g, 87%) as a white foam.
1H-NMR (CDCl3) δ: 0.08 (3H, s), 0.08 (3H, s), 0.90 (9H, s), 1.16–1.33 (3H, m), 1.67–1.76 (2H, m), 1.78–1.87 (2H, m), 1.93 (3H, d, J = 1.0 Hz), 2.15–2.24 (2H, m), 2.43 (1H, dt, J = 13.5, 7.0 Hz), 3.49 (1H, ddd, J = 11.5, 7.0, 4.5 Hz), 4.58 (1H, dd, J = 7.0, 5.0 Hz), 6.06 (1H, t, J = 6.5 Hz), 7.53 (1H, d, J = 1.0 Hz), 8.02 (1H, s); 13C-NMR (CDCl3) δ: −4.9 (s), –4.6 (s), 12.7 (s), 18.1 (s), 21.1 (s), 24.2 (s), 25.9 (s), 29.5 (s), 32.3 (s), 40.3 (s), 72.4 (s), 72.6 (s), 85.2 (s), 88.4 (s), 111.0 (s), 137.6 (s), 150.6 (s), 164.0 (s); IR (KBr) cm−1: 3430, 3170, 3060, 2940, 2860, 2710, 1680, 1470, 1400, 1360, 1290, 1260, 1210; HRMS (MALDI) m/z 433.2129 (Calcd for C20H34N2NaO5Si: 433.2135).
1-[(2R,4S,5R,6S)-4,6-dihydroxy-1-oxaspiro[4.5]decan-2-yl]thymine (6)TBAF (1 M in THF, 0.78 mL, 0.78 mmol) was added to a solution containing 5 (100 mg, 0.24 mmol) in dry THF (5 mL) under N2 atmosphere at 0 °C. The reaction mixture was stirred for 24 h at room temperature, after which it was concentrated in vacuo. The residue (516 mg) was purified by flash column chromatography (CHCl3–MeOH = 10 : 1, followed by MeCN 100%) to yield 6 (30 mg, 42%) as a white foam.
1H-NMR (methanol-d4) δ: 1.28–1.39 (2H, m), 1.47–1.86 (6H, m), 1.88 (3H, d, J = 1.0 Hz), 2.22–2.31 (2H, m), 3.55 (1H, dd, J = 11.5, 4.5, Hz), 4.51 (1H, dd, J = 5.0, 7.0 Hz), 6.23 (1H, t, J = 6.5 Hz), 8.16 (1H, d, J = 1.0 Hz); 13C-NMR (methanol-d4) δ: 11.6 (s), 11.7 (s), 21.5 (s), 24.5 (s), 29.6 (s), 31.8 (s), 40.2 (s), 47.7 (s), 47.9 (s), 48.0 (s), 48.4 (s), 72.4 (s), 83.9 (s), 138.2 (s), 151.7 (s), 165.7 (s); IR (KBr) cm−1: 3480, 3170, 3060, 2960, 2910, 2870, 2790, 1860, 1660, 1510, 1470, 1400, 1380, 1350, 1340, 1280, 1260, 1240, 1210; mp: 209 °C; HRMS (MALDI) m/z 319.1264 (Calcd for C14H20N2NaO5: 319.1270).
1-[(2R,4S,5R,6S)-4-tert-Butyldimethylsilyloxy-6-(4,4′-dimethoxytrityloxy)-1-oxaspiro[4.5]decan-2-yl]thymine (7)DMTrCl (2.50 g, 7.35 mmol) and AgNO3 (1.88 g, 7.35 mmol) were added to a solution of 5 (1.00 g, 2.45 mmol) in the mixed solvent of dry pyridine (5 mL) and dry THF (20 mL) under an N2 atmosphere at room temperature. The reaction mixture was stirred for 3 h at 80 °C. Thereafter, the mixture was filtered by Celite®, and the filtrate was diluted with CHCl3. The solution was washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The residue (4.40 g) was purified via flash column chromatography (n-hexane–AcOEt = 7 : 3) to yield 7 (1.55 g, 88%) as a yellow foam.
1H-NMR (MeCN-d3) δ: −0.17 (3H, s), −0.04 (3H, s), 0.62–0.73 (1H, m), 0.80 (9H, s), 0.95–1.06 (1H, m), 1.34–1.51 (4H, m), 1.58–1.68 (4H, m), 1.73–1.81 (1H, m), 2.20–2.27 (1H, m), 2.41–2.63 (1H, m), 3.01 (1H, dd, J = 11.5, 4.0 Hz), 3.75 (3H, s), 3.76 (3H, s), 4.54 (1H, dd, J = 7.0, 5.0 Hz), 6.12 (1H, t, J = 6.0 Hz), 6.83–6.88 (4H, m), 7.19–7.23 (1H, m), 7.26–7.34 (4H, m), 7.35–7.40 (2H,m), 7.45–7.49 (2H, m), 8.25 (1H, d, J = 1.0 Hz), 9.31 (1H, s); 13C-NMR (MeCN-d3) δ: −4.9 (s), −4.3 (s), 12.5 (s), 18.4 (s), 21.9 (s), 25.0 (s), 26.0 (s), 31.4 (s), 31.6 (s), 42.3 (s), 55.9 (s), 73.0 (s), 77.3 (s), 84.1 (s), 87.7 (s), 90.2 (s), 110.7 (s), 113.8 (s), 127.8 (s), 128.6 (s), 129.0 (s), 131.2 (s), 131.4 (s), 136.8 (s), 137.4 (s), 137.7 (s), 147.7 (s), 151.3 (s), 159.7 (s), 164.9 (s); IR (KBr) cm−1: 3170, 3010, 2930, 2860, 1690, 1610, 1580, 1510, 1460, 1410, 1360, 1340, 1290, 1250, 1220; HRMS (MALDI) m/z 735.3436 (Calcd for C41H52N2NaO7Si: 735.3411).
1-[(2R,4S,5R,6S)-6-(4,4′-Dimethoxytrityloxy)-4-hydroxy-1-oxaspiro[4.5]decan-2-yl]thymine (8)TBAF (1 M in THF, 2.4 mL, 2.4 mmol) was added to a solution of 7 (1.54 g, 2.16 mmol) in dry THF (20 mL) under N2 atmosphere at 0 °C. The reaction mixture was stirred for 12 h at room temperature, after which it was concentrated in vacuo. The residue (2.31 g) was purified by flash column chromatography (CHCl3–AcOEt = 2 : 1) to yield 8 (992 mg, 77%) as a white foam.
1H-NMR (MeCN-d3) δ: 0.58–0.74 (1H, m), 0.93–1.04 (1H, m), 1.32–1.47 (4H, m), 1.56–1.72 (5H, m), 2.18–2.25 (1H, m), 2.35–2.44 (1H, m), 2.83–2.99 (2H, m), 3.73 (3H, s), 3.74 (3H, s), 4.34–4.54 (1H, m), 6.05 (1H, dd, J = 6.5, 5.5 Hz), 6.80–6.86 (4H, m), 7.15–7.21 (1H, m), 7.22–7.27 (2H, m), 7.30–7.39 (4H, m), 7.43–7.48 (2H, m), 8.21 (1H, d, J = 1.0 Hz), 9.22 (1H, s); 13C-NMR (MeCN-d3) δ: 12.7 (s), 21.7 (s), 24.9 (s), 30.7 (s), 31.3 (s), 41.3 (s), 55.8 (s), 71.3 (s), 76.8 (s), 84.0 (s), 87.7 (s), 89.6 (s), 110.6 (s), 113.8 (s), 113.8 (s), 127.7 (s), 128.6 (s), 128.9 (s), 131.1 (s), 131.3 (s), 136.9 (s), 137.5 (s), 137.9 (s), 147.7 (s), 151.5 (s), 159.5 (s), 159.6 (s), 165.2 (s); IR (KBr) cm−1: 3400, 3180, 3060, 3010, 2940, 2860, 2840, 1700, 1610, 1580, 1510, 1460, 1410, 1370, 1280, 1250, 1220; HRMS (MALDI) m/z 621.2571 (Calcd for C 35H38N2NaO7: 621.2577).
1-[(2R,4S,5R,6S)-4-(2-Cyanoethoxy-N,N-diisopropylaminophosphinoxy)-6-(4,4′-dimethoxytrityloxy)-1-oxaspiro[4.5]decan-2-yl]thymine (9)Diisopropylethylamine (0.18 mL, 1.1 mmol) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (87 µL, 0.39 mmol) were added to a solution of 8 (157 mg, 0.26 mmol) in dry CH2Cl2 (3 mL) under N2 atmosphere at 0 °C. The reaction mixture was stirred for 2 h at room temperature. After quenching with sat. NaHCO3 aq. at 0 °C, the whole mixture was diluted with CHCl3. Thereafter, the solution was washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The residue (310 mg) was purified via flash column chromatography (n-hexane–AcOEt = 2 : 1) to yield 9 (186 mg, 88%) as a white foam.
1H-NMR (MeCN-d3) δ: 0.58–0.74 (1H, m), 1.03–1.16 (14H, m), 1.36–1.52 (4H, m), 1.56–1.70 (4H, m), 1.71–1.81 (1H, m), 2.35–2.60 (4H, m), 3.00–3.12 (1H, m), 3.50–3.55 (3H, m), 3.75–3.76 (6H, m), 4.77–4.92 (1H, m), 6.09–6.18 (1H, m), 6.84–6.88 (4H, m), 7.19–7.23 (1H, m), 7.26–7.30 (2H, m), 7.34–7.43 (4H, m), 7.48–7.51 (2H, m), 8.22–8.28 (1H, m), 9.71 (1H, s); 31P-NMR (MeCN-d3) δ: 149.0 (s), 149.2 (s); HRMS (MALDI) m/z 821.3732 (Calcd for C44H55N4NaO8P: 821.3655).
Crystal GrowthFor the X-ray crystallography, 6 was crystallized via vapor diffusion, after which it was dissolved in methanol in the inner vial, which was left open. Next, AcOEt was added to the outer vial, which was capped tightly. The vapors of the outer solvent slowly mixed with the inner solvent, and the crystals of 6 (colorless needle crystals) were produced in the inner vial.
X-Ray CrystallographyA suitable crystal of 6 was carefully selected under an optical microscope and glued to thin glass fibers and mounted on the goniometer in liquid N2 flow. The X-ray diffraction data were collected on a Rigaku R-AXIS XtaLAB P100 diffractometer Mo-Kα radiation. The structure was resolved via the direct method with the SIR-2014 program and refined with the SHELXL program. Further, the structural model was drawn with the ORTEP-3 program. Further information on the crystal structure determinations has been deposited with the Cambridge Crystallographic Data Center (2181435). The data can be obtained free of charge via http://www.ccdc.cam. ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
Syntheses of the OligonucleotidesPhosphoramidite (9) was dissolved in dry THF to a final concentration of 0.1 M. Phosphoramidite (10), dT-phosphoramidite (Sigma), Bz-dC-phosphoramidite (Sigma), and iBu-dG-phosphoramidite (Sigma) were dissolved in dry MeCN to a final concentration of 0.1 M. The oligonucleotides (ON1–11) were synthesized on a 0.2-µmol scale employing an automated DNA synthesizer (Gene Design nS-8 Oligonucleotides Synthesizer) with a 0.25 M Activator 42® in MeCN as an activator. The modified phosphoramidites (9 and 10) were incorporated into the oligonucleotides at a prolonged coupling time, 10 min (cf. 25 s for the coupling of natural phosphoramidites). The oligonucleotides, which were synthesized in the trityl-on mode, were cleaved from the controlled pore glass (GPG) resin via the treatment with 28% aqueous NH3 for 1.5 h at room temperature. All the protecting groups of the oligonucleotides were removed by treatment with 28% aqueous NH3 for 16 h at 55 °C (for ON1–5) and for 8 h at room temperature (for ON8 and ON 10). NH3 was removed in vacuo. The crude oligonucleotides were purified employing a Sep-Pak® Plus C18 cartridge (Waters, U.S.A.) with the removal of the DMTr group during purification employing 1% (v/v) aqueous trifluoroacetic acid. The separated oligonucleotides were further purified via reversed-phase HPLC (Waters XBridge® OST C18 column 2.5 µm, 10 × 50 mm) employing a 0.1 M triethylammonium acetate (TEAA) buffer (pH = 7.0) and MeCN as the eluent. The compositions of the new oligonucleotides (ON2–4, ON7, and ON8) were confirmed by MALDI-TOF/mass spectrometry.
UV Melting ExperimentsFor the UV melting experiments employing the duplexes that were formed by ON1–5, the oligonucleotides were dissolved in 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl to obtain a final concentration of 4.0 µM per strand. The samples were annealed at 100 °C, followed by gradual cooling to room temperature. The melting profiles were recorded at 260 nm from 15 to 75 °C at a scan rate of 0.5 °C/min. The two-point average method was employed to obtain the Tm values, and the final values were determined by averaging three independent measurements, which were accurate within a 1 °C range.
Enzymatic Degradation ExperimentsThe enzymatic degradation experiments were performed employing 1.0 µg/mL CAVP, 10 mM MgCl2, 50 mM Tris–HCl (pH 8.0), and 7.5 µM each oligonucleotide (ON6–11) at 37 °C. Thereafter, the cleavage reaction was performed at 37 °C. A portion of each reaction mixture was removed at timed intervals and heated for 5 min at 100 °C to deactivate the phosphodiesterase. Aliquots of the timed samples were analyzed via reversed-phase HPLC [gradient: 5–15% MeCN in TEAA buffer (0.1 M, pH 7.0) for 20 min, flow rate: 1.0 mL/min, column temperature: 50 °C] to evaluate the amount of intact oligonucleotide remaining. The percentage of intact oligonucleotide in each sample was calculated and plotted against the digestion time to obtain the degradation curve in time.