Results and Discussion
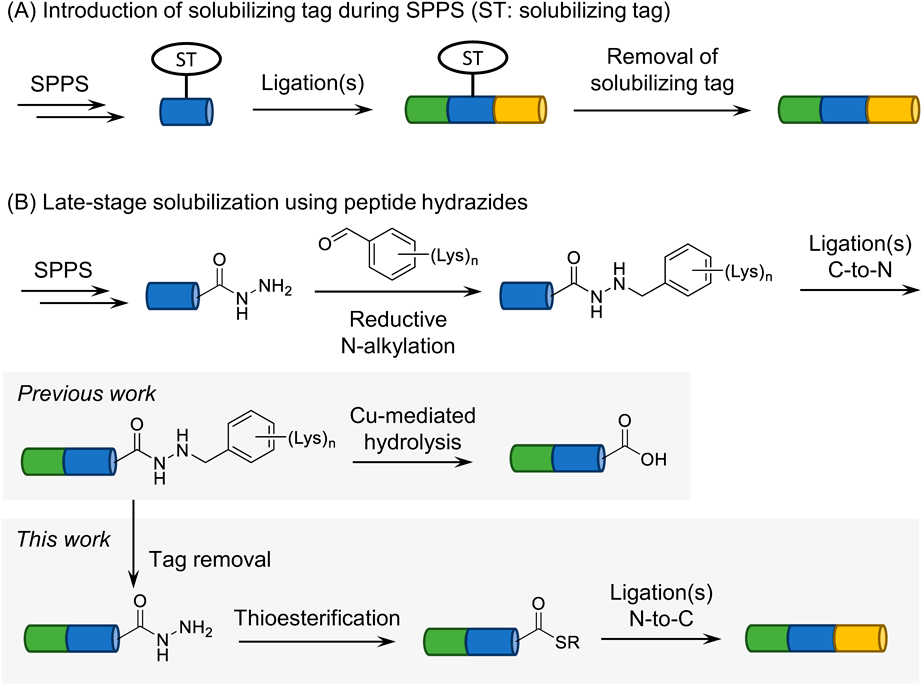
N-Alkylation of Peptide Hydrazides with Different Substituents on the Benzene RingWe envisioned the reaction sequence in Chart 1B for the late-stage solubilization of peptide hydrazides followed by regeneration of a free hydrazide to permit its use as a handle in further modifications. Benzyl-type protecting groups on a heteroatom are usually removed in peptide synthesis by treatment with trifluoroacetic acid (TFA), and the substituents on the benzene ring can tune the reactivity. TFA deprotection would be useful in the subsequent conversion to the thioester because a protocol has been developed to activate a hydrazide with the aid of NaNO2 in TFA which affords an acyl azide, which can react with a thiol to yield the corresponding thioester.31)
In our initial experiments, reductive N-alkylation of peptide hydrazide with benzaldehydes bearing different substituents was performed under the condition used previously30) (Table 1). Ac-LYRAG-NHNH2 (1), a peptide hydrazide that has been widely used as a model sequence was reacted with 4-anisaldehyde (2a) or 2,4-dimethoxybenzaldehyde (2b) in acetic acid and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (50% AcOH-HFIP) for 1 h.32) Then, 20 equivalents of 2-picoline-borane complex (pic-BH3) was added to the reaction mixture and reacted for an additional 1 h, affording the desired N-alkyl hydrazide (3a, 3b) in 86% or 85% yield, respectively (entries 1 and 3). The same reaction proceeded even in 10 min to achieve reduction with high yields (entries 2 and 4). The reaction with 2,4,6-trimethoxybenzaldehyde (2c) afforded the N-alkyl hydrazide (3c) in 62% yield and 29% starting material remained after 1 h reaction whereas a shorter reaction time gave the product in higher yield (entries 5–7), suggesting that this may result from decomposition of the generated N-alkyl hydrazide under the slightly acidic reaction conditions. Indeed, the free hydrazide (1) was formed within 5 min when the isolated 3c was treated with 50% AcOH-HFIP solution (Supplementary materials, Fig. S6).
Table 1. Reductive
N-Alkilation of a Peptide Hydrazide
a)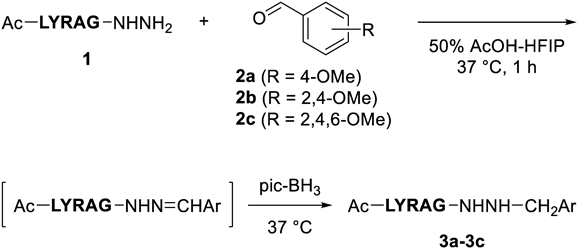 |
|---|
| Entry | Aldehyde | Reduction time [min] | HPLC purity of 3 [%]b) |
|---|
| 1 | 2a | 60 | 86 |
| 2 | 10 | 88 (49)c) |
| 3 | 2b | 60 | 85 |
| 4 | 10 | 89 (48)c) |
| 5 | 2c | 60 | 62 |
| 6 | 10 | 88 |
| 7 | 5 | 87 (41)c) |
a) Reactions were performed as follows: The peptide hydrazide 1 (1 mM) and aldehyde (2a–2c, 1.5 mM) were dissolved in 50% AcOH-HFIP and then incubated at 37 °C for 1 h. After the addition of pic-BH3 (20 equivalents) to the mixture, the reduction was performed at 37 °C. After removal of the solvent in an air stream, the crude material was washed with Et2O. The resulting precipitate was analyzed by HPLC. b) Detected at 220 nm. c) Isolated yield.
We then examined the reactivity and stability of N-alkyl hydrazides (Table 2). Deprotection of the monomethoxybenzyl derivative (3a) by TFA and triisopropylsilane (TIS) as a carbocation scavenger33) did not occur within 1 h while the di- or trimethoxybenzyl derivatives (3b and 3c) were deprotected giving the free hydrazide (entries 1, 2 and 6). This trend can be explained in terms of the electron-donating ability of the benzene ring increasing the reactivity in acidic conditions as a result of the enhanced stability of the benzyl cation formed by the cleavage.34)
Table 2. Reactivity and Stability of
N-Alkyl Hydrazide Peptides
 |
|---|
| Entry | Substrate | Conditions | HPLC purity [%]a) |
|---|
| 3 | 1 |
|---|
| 1 | 3a | TFA-TIS (95 : 5)b) | 98 | <1 |
| 2 | 3b | TFA-TIS (95 : 5) | <1 | 87 |
| 3 | NCLc) | 88 | <1 |
| 4 | Thz deprotectiond) | 97 | <1 |
| 5 | Desulfurizatione) | 81 | 3 |
| 6 | 3c | TFA-TIS (95 : 5) | <1 | 88 |
| 7 | NCL | 72 | 3 |
| 8 | Thz deprotection | 88 | 7 |
| 9 | Desulfurization | 63 | 12 |
a) Detected at 220 nm. b) TFA-TIS (95 : 5, v/v), 37 °C, 1 h. c) 6 M Gn·HCl, 0.2 M phosphate, 100 mM MPAA, 50 mM TCEP·HCl, pH 7.1, 37 °C, 12 h. d) 6 M Gn·HCl, 0.2 M phosphate, 100 mM MPAA, 50 mM TCEP·HCl, 200 mM MeONH2·HCl, pH 5.1, 37 °C, 12 h. e) 6 M Gn·HCl, 0.2 M phosphate, 250 mM TCEP·HCl, 150 mM MESNa, 50 mM VA-044, pH 6.2, 37 °C, 12 h.
These benzyl moieties must remain attached during the protein synthesis reactions that include NCL, ring-opening of a thiazolidine (Thz) ring to be used in cysteine protection35) or desulfurization of cysteine (Cys) to enable peptide assembly at an alanine site.36) Dimethoxybenzyl hydrazide (3b) is stable in NCL reactions, Thz deprotection, and desulfurization conditions (entries 3–5), whereas hydrazides such as 3c with a trimethoxybenzyl moiety are relatively unstable, and liable to decompose (entries 7–9). In light of these results, we decided in this study to use a dialkoxybenzyl linker to solubilize peptide hydrazides.
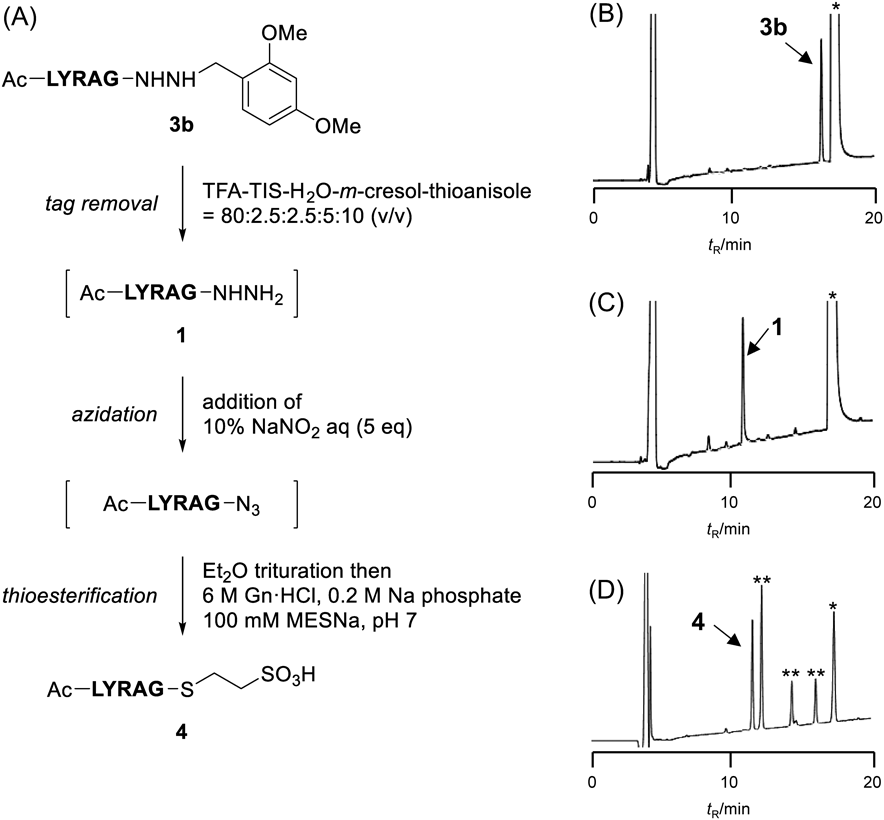
One-Pot Reaction for Tag Removal and Subsequent Azidation and ThiolysisWe attempted a one-pot conversion of the tag-linked hydrazide to the corresponding thioester which can be used as a handle in further modifications (Fig. 1). A peptide hydrazide with a dimethoxybenzyl linker (3b) was treated with a TFA-based deprotection cocktail (TFA-TIS-H2O-m-cresol-thioanisole = 80 : 2.5 : 2.5 : 5 : 10, v/v), which is an optimized condition for acyl azide formation,31) and the progress of the reaction was monitored by HPLC. The cleavage of the benzyl linker proceeded smoothly within 1 h to give the free hydrazide (1) (Figs. 1B, 1C). An aqueous solution of NaNO2 (5 equivalents) was added to the resulting mixture, leading to the peptidyl azide intermediate. After a 20 min reaction at −10 °C followed by trituration with cold Et2O, the precipitate was dissolved in a buffer containing sodium 2-mercaptoethanesulfonate (MESNa). HPLC analysis of the thiolysis reaction mixture indicated that the desired thioester (4) had been formed without any significant amount of byproducts (Fig. 1D).
Synthesis of a Lys63-Linked Di-Ubiquitin DerivativeA synthetic application of our strategy was confirmed through the synthesis of Lys63-linked di-ubiquitin (Ub), which is one of the Ub linkage types found in many cellular processes.37) Brik and colleagues reported the chemical synthesis of SUMO-2 linked to Lys63-di-Ub.38) They encountered synthetic difficulties due to the poor solubility of a synthetic intermediate, the Lys63-di-Ub thioester derivative. To address this problem, we used the method shown in Chart 2 to synthesize a Lys63-linked di-Ub derivative containing biotin.
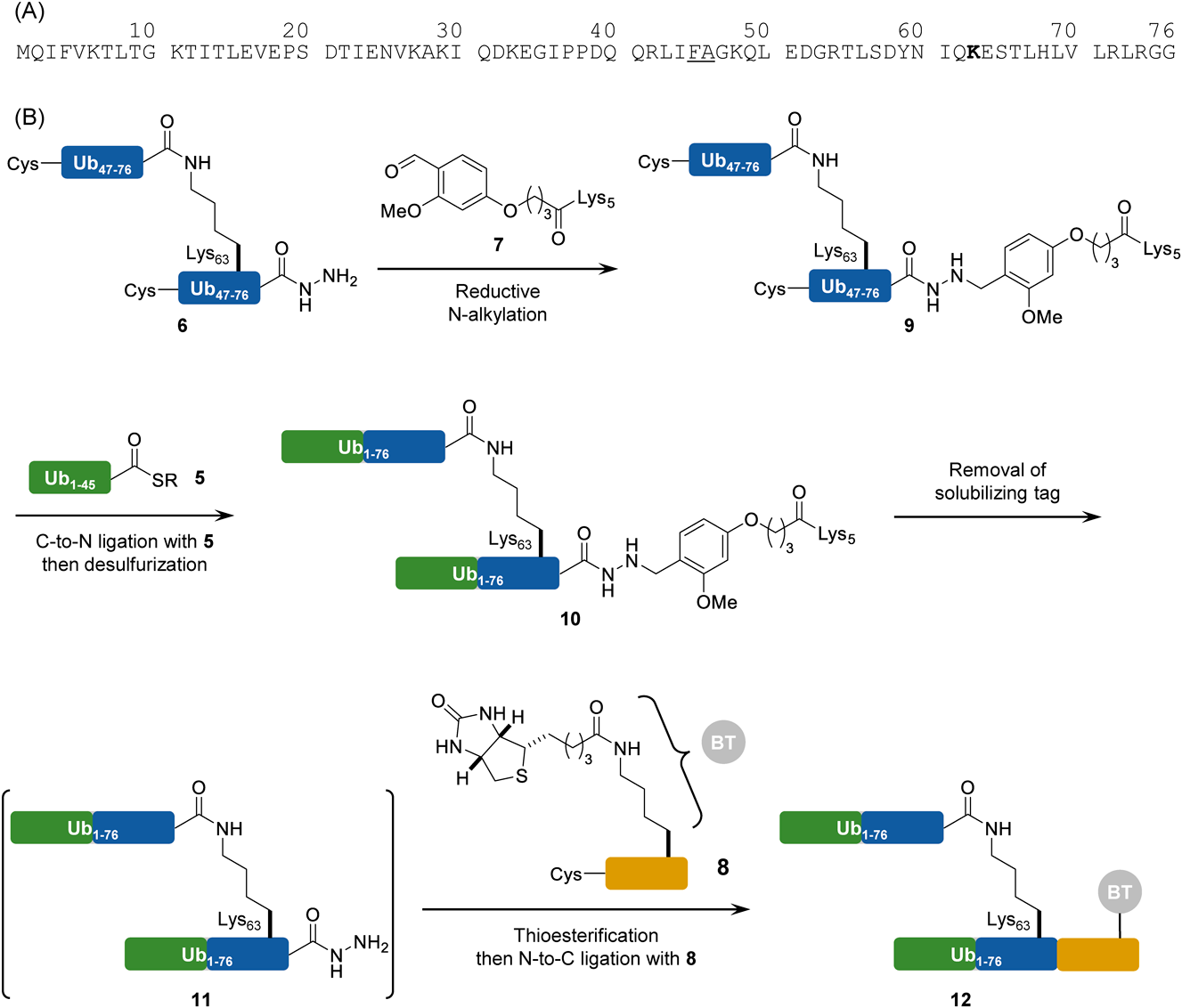
The sequence of Ub was disconnected to two peptide segments, Ub1-45 and Ub46-76, and the Ala46 was replaced by Cys to join these two peptides through an NCL reaction. The C-terminal segment can be synthesized as a branched form during SPPS. The Lys63-linked di-Ub46-76 (6) as a hydrazide form should combine with a solubilizing tag possessing a dialkoxybenzyl linker (7) through reductive N-alkylation. A NCL reaction between two peptide segments (5 and 9) followed by desulfurization will afford Lys63-linked di-Ub1-76 (10) containing the solubilizing tag. After removal of the tag and subsequent conversion of the alkyl hydrzide to a thioester, NCL of the resulting peptide with a biotin-containing peptide (8) that has Cys at the N-terminus should give the desired di-Ub derivative (12).
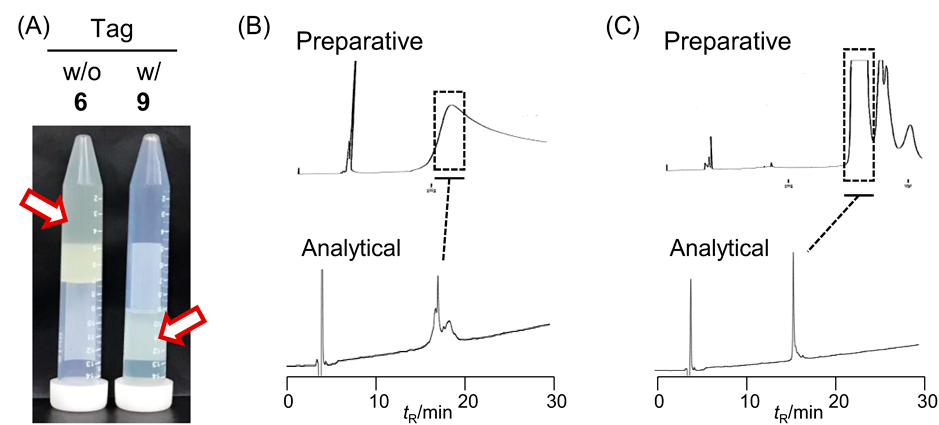
Peptide syntheses covering the Ub sequence were performed on a Trt ChemMatrix resin bound to hydrazine. An N-(2,4-dimethoxybenzyl)-glycine unit was introduced at Gly53 to prevent aspartimide formation during the SPPS.39) Pseudoproline units at the Leu8-Thr9, Ile13-Thr14, Leu56-Ser57, and Ser65-Thr66 were used to improve the synthetic efficiency.40) The N-terminal segment Ub1-45 was synthesized as the hydrazide and converted to the MESNa thioester, affording the Ub1-45 thioester (5). The C-terminal segment was prepared as the hydrazide, and the branched structure at Lys63 was formed during SPPS using the orthogonal building block Lys(Alloc) at this position. After elongation of the Boc-Ub46-76Ala46Cys peptide, the Alloc group at Lys63 was removed by reaction with Pd(PPh3)4 and PhSiH3, and the additional Ub46-76Ala46Cys sequence was elongated at the deprotected Lys63 amino group to yield the branched peptide hydrazide (6). Although the N-terminal segment 5 was obtained successfully, the branched C-terminal segment 6 formed a gel when dissolving the crude material obtained after global deprotection in 40% aqueous CH3CN containing 0.1% TFA (Fig. 2A). The solvent was able to dissolve the crude material but imperfect peak separation was observed during HPLC purification (Fig. 2B), causing multiple purification steps and ultimately, an isolated yield of only 0.2%. Consequently, we tried to introduce the solubilizing tag prior to the purification steps.
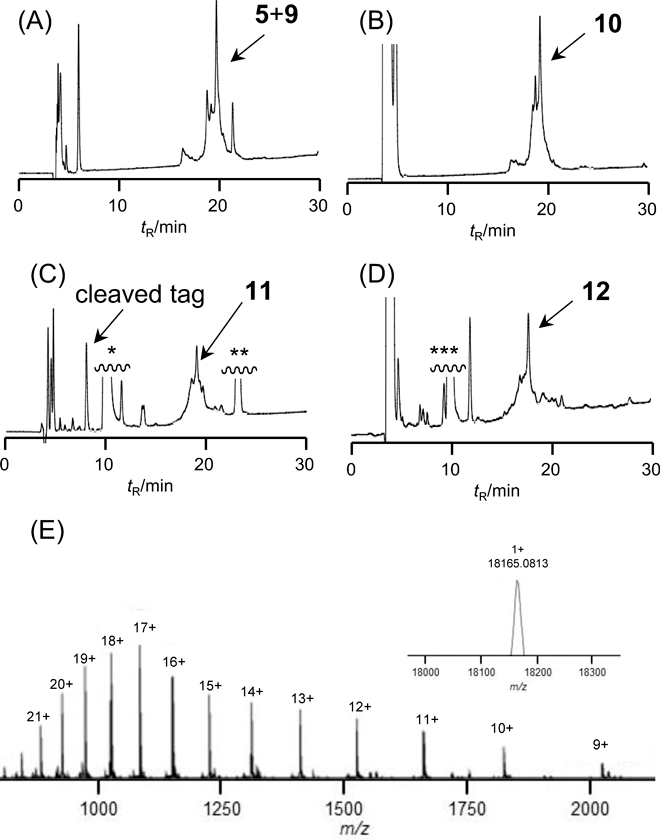
The benzaldehyde derivative (13) was synthesized as shown in Chart 3. Alkylation of 4-formyl-3-methoxyphenol with ethyl 4-bromobutyrate followed by hydrolysis afforded the necessary formyl compound, 4-(4-formyl-3-methoxyphenoxy)butanoic acid (13) in 66% overall yield. As a hydrophilic moiety, Lys 5-mer was coupled with 13 on a Rink Amide AM resin, and this was followed by global deprotection, giving the solubilizing tag (7). The reductive N-alkylation of the hydrazide (6) as a crude material with the solubilizing tag (7) was performed in 50% AcOH-HFIP containing pic-BH3 for 10 min to produce the tag-containing segment (9).41) The resulting crude material showed improved solubility in 40% aqueous CH3CN containing 0.1% TFA without gel formation, leading to good peak separation in the HPLC purification (Fig. 2C). The isolated yield of segment 9 was 2%, ten times higher than the yield obtained in the absence of the solubilizing tag. Methyl thioglycolate-mediated NCL between 5 and 9 followed by desulfurization was performed in a one-pot reaction,42) yielding the full-length protein containing the solubilizing tag (10) in 26% isolated yield without any difficulty associated with poor solubility (Fig. 3). The reaction sequence containing the removal of the solubilizing tag and azide formation by NaNO2 in TFA resulted in a conversion of the alkyl hydrazide to the corresponding peptidyl azide. After being washed with Et2O to remove excess scavengers, the resulting precipitate was subjected to modification with the biotin-containing peptide H-CLPLTGGGK(biotin)-NH2 (8) by NCL in a one-pot manner, yielding the Lys63-linked di-Ub with a biotin label from the tag-containing segment (10) in 18% isolated yield.
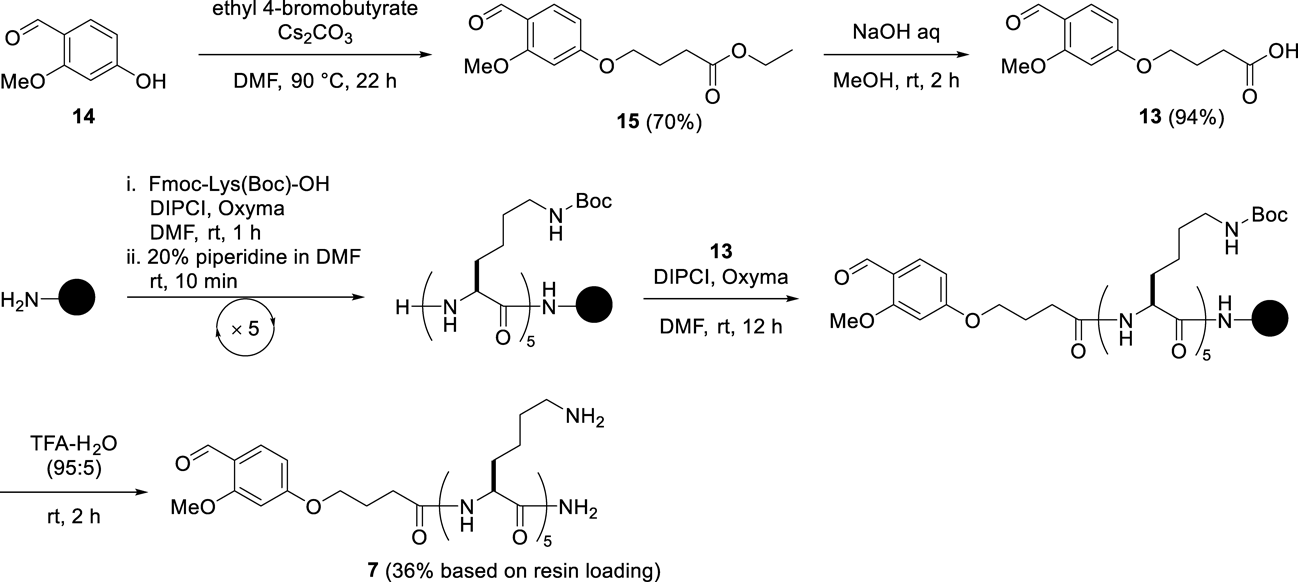
Chart 3. Synthesis of Solubilizing Tag 7
Experimental
General InformationAll commercially available reagents and protected amino acids were used without further purification. Thin-layer chromatography was performed on Merck 60F254 precoated silica gel plates which were visualized by fluorescence quenching under UV light and by staining with 2,4-dinitrophenylhydrazine. Column chromatography was carried out using a silica gel 60 N (Kanto Chemical, Tokyo, Japan).
1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were recorded using a Bruker AVANCE III HD spectrometer. Chemical shifts are reported in δ (ppm) relative to tetramethylsilane as an internal standard. Mass spectra were recorded on a Bruker Compact (ESI-Q-TOF) mass spectrometer. Melting points were measured using a CORNES Technologies MPA 100. IR spectra were recorded on a JASCO FT/IR 6300 with ATR and are reported in wavenumbers (cm−1).
HPLC analyses were carried out at 30 °C on a system using JASCO PU-2089 Plus, UV-2070 Plus, AS-2057 Plus, and CO-2060 Plus. A solvent system consisting of 0.1% TFA aqueous solution (v/v, solvent A) and 0.1% TFA in CH3CN (v/v, solvent B) was used for HPLC elution. Characterization data of peptides are summarized in Table 3.
Table 3. Peptide Characterization Data
| Comp. # | Yield (%) | HPLC | MS (m/z) |
|---|
| Analytical | Preparative |
|---|
| Gradient (%) (column used)a) | Retention time (min) | Gradient (%) (column used)b) | Calcd | Found |
|---|
| 1 | 50 | 10–60 (C8)c) | 10.2 | 10–20 (C8)c) | 635.4 [M + H]+ | 635.4 |
| 3a | 49 | 15–50 (C8)c) | 13.2 | 20–30 (C8)c) | 755.4 [M + H]+ | 755.4 |
| 3b | 48 | 15–50 (C8)c) | 13.6 | 20–30 (C8)c) | 785.4 [M + H]+ | 785.4 |
| 3c | 41 | 15–50 (C8)c) | 14.3 | 20–30 (C8)c) | 815.4 [M + H]+ | 815.4 |
| 4 | — | 10–40 (C8)d) | 11.6 | — | 745.3 [M + H]+ | 745.3 |
| 5 | 12 | 10–60 (PR)c) | 20.3 | 27–37 (PR)e) | 1309.7 [M + 4H]4+ | 1309.7 |
| 6 | 0.2 | 10–60 (PR)c) | 18.9 | 27–37 (PR)e) | 1399.7 [M + 5H]5+ | 1399.5 |
| 7 | 36 | 5–35 (C18)c) | 16.1 | 10–20 (C18)c) | 878.6 [M + H]+ | 878.6 |
| 8 | 19 | 10–60 (C18)c) | 13.9 | 18–28 (C18)c) | 1070.5 [M + H]+ | 1070.6 |
| 9 | 2 | 10–60 (PR)c) | 18.4 | 27–37 (PR)e) | 1572.3 [M + 5H]5+ | 1572.1 |
| 10 | 26 | 20–50 (PR)c) | 19.3 | 28–38 (PR)e) | 1199.5 [M + 15H]15+ | 1199.8 |
| 11 | — | 25–45 (PR)c) | 19.1 | — | 1142.0 [M + 15H]15+ | 1142.2 |
| 12 | 18 | 25–45 (PR)c) | 17.8 | 28–38 (PR)e) | 1211.3 [M + 15H]15+ | 1211.4 |
a) C8: Cosmosil 5C8-AR-300 analytical column; C18: Cosmosil 5C18-AR-II analytical column; PR: Cosmosil Protein-R analytical column. b) C8: Cosmosil 5C8-AR-300 preparative column; C18: Cosmosil 5C18-AR-II preparative column; PR: Cosmosil Protein-R semi-preparative column. c) Gradient over 30 min. d) Gradient over 20 min. e) Gradient over 60 min.
Side chain protecting groups of Fmoc amino acids are as follows: Boc: H and K; Ot-Bu: D and E; Pbf: R; t-Bu: S, T and Y; Trt: C, N and Q; Alloc: K for K63 of proximal Ub or K9 of peptide 8. Fmoc-(Dmb)Gly-OH was prepared by a previously published procedure.43) Fmoc SPPS was performed with an automated microwave-assisted synthesis or a manual synthesis. The microwave-assisted SPPS using a Biotage Initiator+ Alstra peptide synthesizer with a 10 mL open-type vial was conducted as follows: coupling with amino acids except for Cys(Trt) or Arg(Pbf): Fmoc-protected amino acid, ethyl 2-cyano-2-(hydroxyamino)acetate (Oxyma), and N,N′-diisopropylcarbodiimide (DIPCI) (4.0 equivalents each) in N,N-dimethylformamide (DMF) (0.5 M), 5 min, 75 °C under microwave irradiation; coupling with Cys(Trt) or Arg(Pbf): Fmoc-protected amino acid, Oxyma and DIPCI (4.0 equivalents each) in DMF (0.5 M), 60 min, room temperature (r.t.); Fmoc removal: 20% (v/v) piperidine in DMF, 3 min, 50 °C under microwave irradiation. The reaction temperatures were monitored by an IR sensor. Except where specifically noted, the manual synthesis was conducted as follows: 1. (coupling) Fmoc-protected amino acid, Oxyma, and DIPCI (4.0 equivalents each) in DMF (0.3 M), 60 min, r.t.; 2. (Fmoc removal) 20% (v/v) piperidine in DMF, 10 min, r.t..
Deprotection of Alloc Group on Solid SupportThe peptidyl resin was swollen in dichloromethane (DCM) and solutions of Pd(PPh3)4 (0.24 equivalents), then PhSiH3 (24 equivalents) in DCM were added to the reaction mixture which was then stirred at r.t. under an argon atmosphere for 2 × 30 min. After filtration, the resulting resin was washed three times each with the following solvents: tetrahydrofuran (THF), DMF, DCM, 0.5% (v/v) N,N-diisopropylethylamine (DIPEA) in DCM, 0.02 M sodium diethyldithiocarbamate in DMF, DMF and DCM.44)
Preparation of Hydrazide-Incorporated ResinTrityl OH-ChemMatrix resin was swollen in dry DCM and treated with thionyl chloride (10 equivalents) at r.t. for 2 h. After being washed with dry DCM and dry DMF, the resulting resin was again swollen with dry DCM. A solution of 9-fluorenylmethyl carbazate (4.0 equivalents) and DIPEA (10 equivalents) in dry DMF was added dropwise to the resin at 0 °C. The reaction mixture was shaken at r.t. overnight, then MeOH was added to cap extra sites. After 10 min, the resin was filtered and washed successively with DMF, water, DMF, MeOH and DCM. The loading was confirmed by quantification of the Fmoc group.
Examinations Using Model PeptidesSynthesis of Model Peptide Hydrazide 1Model peptide Ac-LYRAG-NHNH21 was elongated on the hydrazine-incorporated resin by the microwave-assisted synthesis and the resulting resin was treated with TFA-TIS-H2O (95 : 2.5 : 2.5 (v/v), 50 µL/1 mg resin) at r.t. for 2 h. The resin in the reaction mixture was removed by filtration and then the filtrate was concentrated in a stream of air. Cold Et2O was added to the resulting filtrate to give a precipitate. After having been repeatedly washed with Et2O, the precipitated crude peptides were purified by preparative HPLC.
Reductive N-Alkylation of a Peptide HydrazideThe peptide hydrazide 1 (0.10 µmol) was reacted with aldehyde 2a, 2b or 2c (1.5 equivalents) in 50% (v/v) AcOH-HFIP (100 µL) at 37 °C. After 1 h reaction, pic-BH3 (20 equivalents) was added, and the mixture was incubated at 37 °C. After the appropriate time shown in Table 1, TFA (2 µL) was added to the reaction mixture. The resulting solution was concentrated in a stream of air and then washed with Et2O. The precipitate that was formed was dissolved in 50% (v/v) aqueous CH3CN (100 µL) and the solution was analyzed by HPLC. For isolation, hydrazide 1 (5.0 µmol) was used and the product was purified by preparative HPLC.
Examination of Reactivity and Stability of N-Alkyl Hydrazide PeptidesThe N-alkyl hydrazide (3a–3c, 0.10 µmol) was dissolved in each of the following solutions and incubated at 37 °C for the appropriate time shown in Table 2. 1. (Tag removal condition) 5% (v/v) TIS-TFA (100 µL); 2. (NCL condition) 6 M guanidine (Gn)·HCl, 0.2 M Na2HPO4, 100 mM 4-mercaptobenzoic acid (MPAA), 50 mM tris(2-carboxyethyl)phosphine (TCEP)·HCl, pH 7.1; 3. (Thz deprotection) 6 M Gn·HCl, 0.2 M Na2HPO4, 100 mM MPAA, 50 mM TCEP·HCl, 200 mM MeONH2·HCl, pH 5.1; 4. (Desulfurization) 6 M Gn·HCl, 0.2 M Na2HPO4, 150 mM MESNa, 250 mM TCEP·HCl, 50 mM 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044), pH 6.2.
One-Pot Conversion of Alkyl Hydrazides to ThioestersThe-N-alkyl hydrazide (3b, 0.10 µmol) was treated with TFA-TIS-H2O-m-cresol-thioanisole (80 : 2.5 : 2.5 : 5 : 10, (v/v), 0.1 µmol/100 µL) and the mixture was allowed to react for 1 h at 37 °C. An aqueous solution of NaNO2 (5 µL, 5 equivalents) was added to the mixture at −10 °C, and the solution was incubated at −10 °C for 20 min. To the reaction mixture was added cold Et2O to give a precipitate which was collected by centrifugation and thoroughly washed with Et2O to afford a peptide azide. The crude product was dissolved in 100 µL of buffer (6 M Gn·HCl, 0.2 M Na phosphate, 100 mM MESNa, pH 7.0), and the solution was incubated at 37 °C to yield the thioester (4).
Synthesis of the Solubilizing TagCompound 154-Formyl-3-methoxyphenol (14, 0.50 g, 3.3 mmol) was dissolved in DMF (2 mL), then Cs2CO3 (0.76 g, 3.9 mmol) was added in one portion followed by the addition of neat ethyl 4-bromobutyrate (0.57 mL, 3.9 mmol). The temperature was then increased to 90 °C and maintained there for 22 h and then cooled to r.t. and filtered. The filtrate was concentrated under reduced pressure. The residue was dissolved in EtOAc (20 mL), the organic phase was washed with aqueous 1 M HCl (3 × 5 mL) and aqueous 1 M NaOH (3 × 1 mL), and then dried over Na2SO4. After filtration and subsequent removal of the solvent under reduced pressure, chromatographic purification on silica gel (n-hexane/EtOAc = 3 : 1, (v/v)) gave compound 15 in 70% isolated yield (0.61 g) as a white solid. 1H-NMR (CDCl3) δ: 1.27 (3H, t, J = 7.1 Hz), 2.14 (2H, tt, J = 7.2, 6.2 Hz), 2.52 (2H, t, J = 7.2 Hz), 3.9 (3H, s), 4.09 (2H, t, J = 6.2 Hz), 4.16 (2H, q, J = 7.1 Hz), 6.45 (1H, d, J = 2.2 Hz), 6.53 (1H, dd, J = 8.7, 2.2 Hz), 7.79 (1H, d, J = 8.7 Hz), 10.29 (1H, s); 13C-NMR (CDCl3) δ: 14.2, 24.4, 30.6, 55.6, 60.6, 67.2, 98.3, 106.3, 119.1, 130.8, 163.6, 165.4, 173.0, 188.3; mp: 47 °C; IR (ATR) cm−1: 1730 (CO), 1600 (CO); ESI-MS m/z: 289.0959 (Calcd for C14H18NaO5: 289.1046).
Compound 13Compound 15 (0.61 g, 2.3 mmol) in MeOH (10 mL) was treated with 2 M NaOH aqueous solution (2.3 mL). After being stirred at r.t. for 2 h, the reaction mixture was concentrated in vacuo to obtain a crude material. A solution of this material in H2O (1 mL) was acidified with aqueous 6 M HCl to pH 1–2 to afford a precipitate which was collected by filtration, washed with H2O, and dried to give compound 13 in 94% isolated yield (0.51 g) as a white solid. 1H-NMR (DMSO-d6) δ: 1.97 (2H, tt, J = 7.3, 6.5 Hz), 2.4 (2H, t, J = 7.3 Hz), 3.91 (3H, s), 4.12 (2H, t, J = 6.5 Hz), 6.64 (1H, dd, J = 8.6, 2.1 Hz), 6.69 (1H, d, J = 2.1 Hz), 7.65 (1H, d, J = 8.6 Hz), 10.17 (1H, s), 12.19 (1H, s); 13C-NMR (DMSO-d6) δ: 24.5, 30.4, 56.5, 67.8, 99.1, 107.6, 118.6, 130.3, 163.9, 165.8, 174.5, 187.7; mp: 130 °C; IR (ATR) cm−1: 3523 (OH), 1731 (CO), 1588 (CO), 1174 (CO); ESI-MS m/z: 237.0909 (Calcd for C12H13O5: 237.0768).
SPPS for Solubilizing Tag 7The Lys 5-mer was elongated on a Rink Amide AM resin (0.28 g, 0.20 mmol, 0.70 mmol/g) using a manual protocol to produce a protected peptidyl resin. After removal of the final Fmoc group, compound 13 (0.19 g, 0.80 mmol) was coupled in the presence of DIPCI (4 equivalents) and Oxyma (4 equivalents) in DMF. The coupling proceeded to completion within 12 h. Cleavage of the solubilizing tag from the resin was performed using TFA–H2O (95 : 5 (v/v), 50 µL/1 mg resin) at r.t. for 2 h. The resin in the reaction mixture was filtered off and then the filtrate was concentrated in a stream of air. The crude material was dissolved in 0.1% TFA-containing H2O (3 mL) and washed with Et2O (3 × 5 mL). This was purified by preparative HPLC.
Synthesis of Lys63-Linked Di-Ub DerivativeSynthesis of Biotin Peptide 8Fmoc-Lys(Alloc)-OH was coupled to Rink Amide ChemMareix resin (0.42 mmo/g, 0.12 g, 0.05 mmol) and the peptide was elongated using microwave-assisted Fmoc SPPS. Alloc group was deprotected as described above and then biotin (48 mg, 0.20 mmol, 4 equivalents) was coupled to the resulting amino group using 1-[bis(dimethylamino)methylene]- 1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate, (HATU) (72 mg, 0.19 mmol, 3.8 equivalents) and DIPEA (70 µL, 0.40 mmol, 8 equivalents) in 50% (v/v) DMSO–DMF for 30 min at r.t. Cleavage of the peptide was performed using TFA–TIS–H2O (95 : 2.5 : 2.5, (v/v), 50 µL/1 mg resin) at r.t. After 2 h cleavage, the reaction mixture was filtrated off and then the filtrate was concentrated in a sream of N2. Cold Et2O was added to the resulting filtrate to give a precipitate. After successive washing with Et2O, the crude product was purified by preparative HPLC.
Synthesis of the Peptide Thioester (5)On hydrazine incorporated resin (0.31 mmol/g), the peptide was elongated using microwave-assisted Fmoc SPPS. Fmoc-Leu-Thr(ΨMe,Mepro)-OH and Fmoc-Ile-Thr(ΨMe,Mepro)-OH were used for the Leu8-Thr9 and Ile13-Thr14 sites. The resin was treated with TFA–TIS–H2O-m-cresol-thioanisole (80 : 2.5 : 2.5 : 5 : 10, (v/v), 50 µL/1 mg resin) at r.t. After 2 h, 10% (w/w) NaNO2 aq (1 µL/1 mg resin) was added to the mixture at −10 °C and the solution was incubated at −10 °C for 20 min. To the reaction mixture was added cold Et2O giving a precipitate. The precipitate was collected by centrifugation and thoroughly washed with Et2O to afford a crude peptide azide. The crude product was dissolved into 5% (w/w) MESNa in buffer (6 M Gn·HCl, 0.2 M Na phosphate, pH 7.3, 5 mL). After 30 min at r.t., the solution was acidified to pH 2–3 by the addition of TFA and purified by preparative HPLC.
Synthesis of Peptide Hydrazides 6 and 9Peptide 6 was elongated on a hydrazine incorporated resin (338 mg, 0.10 mmol, 0.31 mmol/g) using a manual SPPS procedure. On this resin, standard Fmoc SPPS (coupling: 3.0 equivalents of amino acid using HATU (2.8 equivalents) and DIPEA (6.0 equivalents) in DMF, 10 min, r.t.; Fmoc removal: 20% (v/v) piperidine/DMF, 10 min, r.t.) was performed for the chain elongation to give a protected peptide resin. Fmoc-Leu-Ser(ΨMe,Mepro)-OH, Fmoc-Ser(t-Bu)-Thr(ΨMe,Mepro)-OH and Fmoc-(Dmb)Gly-OH were used for the Leu56–Ser57, Ser65–Thr66 and Gly53 sites, respectively. After elongation of the Boc-Ub46-76Ala46Cys peptide, the Alloc group at Lys63 was removed. The remaining sequence was further elongated at the resulting Lys side chain. The resulting resin (666 mg, 50 µmol) was treated with TFA–TIS–H2O (95 : 2.5 : 2.5 (v/v), 50 µL/1 mg resin) at r.t. for 2 h. After filtration of the resin in the reaction mixture, the filtrate was removed in an air stream and the addition of cooled Et2O to the residue afforded a precipitate. After successively washed with Et2O, the crude peptide was purified by preparative HPLC. Four times purification was required to attain a pure product 6 due to the low peak separation.
The crude material obtained from cleavage of the resin (662 mg, 50 µmol) was dissolved in 50% AcOH–HFIP (10 mL). The solubilizing tag 7 (110 mg, 73 µmol, 1.5 equivalents) was added, and the mixture was allowed to react for 2 h at 37 °C. Following the addition of Pic–BH3 (100 mg, 970 µmol, 20 equivalents), the mixture was incubated for additional 10 min at 37 °C. After the addition of TFA (370 µL) to the solution, the solvent was removed in an air stream, affording the crude material (9). The residue was dissolved in H2O-CH3CN (5 : 3 (v/v), 8 mL) and purified by preparative HPLC. In this case two purification steps were required to purify the product (9).
NCL between 5 and 9 Followed by DesulfurizationThe N-Cys peptide (9, 0.90 µmol, 1.0 equivalents) and peptide thioester (5, 1.85 µmol, 2.05 equivalents) were dissolved in 900 µL of ligation buffer (6 M Gn·HCl, 0.2 M Na phosphate, 50 mM methyl thioglycolate, 30 mM TCEP·HCl, pH 7.3), and the solution was incubated at 37 °C. After completion of the ligation in 3 h, desulfurization buffer (6 M Gn·HCl, 0.2 M Na phosphate, 500 mM TCEP·HCl, 200 mM MESNa, pH 6.6, 900 µL) and VA-044 (31 mg, 90 µmol, 100 equivalents) as a solid were added. The mixture was allowed to react at 37 °C for 18 h. After pH adjustment to 1–2 by addition of TFA, the desulfurized product (10) was purified by preparative HPLC.
Tag Removal from 10 Followed by NCL with Peptide 8The peptide (10, 0.23 µmol) was treated with TFA–TIS–H2O-m-cresol-thioanisole (80 : 2.5 : 2.5 : 5 : 10, (v/v), 0.1 µmol/100 µL) and the mixture was allowed to react for 3 h at 37 °C. Then, NaNO2 aq (5 µL, 5 equivalents) was added at −10 °C, and the solution was incubated at −10 °C for 30 min. Cold Et2O was added to the reaction mixture to give a precipitate which was collected by centrifugation and thoroughly washed with Et2O to afford a peptide azide. The crude product was dissolved in 400 µL of ligation buffer (6 M Gn·HCl, 0.2 M Na phosphate, 100 mM MPAA, pH 7.3) containing peptide 8 (0.46 µmol, 2 equivalents), and the solution was incubated at 37 °C. The reaction was monitored by HPLC and essentially completed within 5 h. After adjustment to pH 1–2 by addition of TFA, the ligated product (12) was isolated and purified by preparative HPLC.