Results and Discussion
The leaves of C. cascarilloides were extracted with MeOH, and the MeOH extract was separated via liquid-liquid partition. The EtOAc fractions thus obtained were separated using various chromatographic techniques to isolate seven new compounds (1–7) and nine known ones (8–16).
The structures of the new compounds were mainly elucidated using one-dimensional NMR spectroscopies (1H-, 13C-NMR, and the distortion-less enhancement of polarization transfer spectra), together with two-dimensional [1H–1H correlation spectroscopy (COSY), heteronuclear single-quantum correlation spectroscopy (HSQC), heteronuclear multiple-bond correlation spectroscopy (HMBC)] NMR experiments and other spectroscopic evidence.
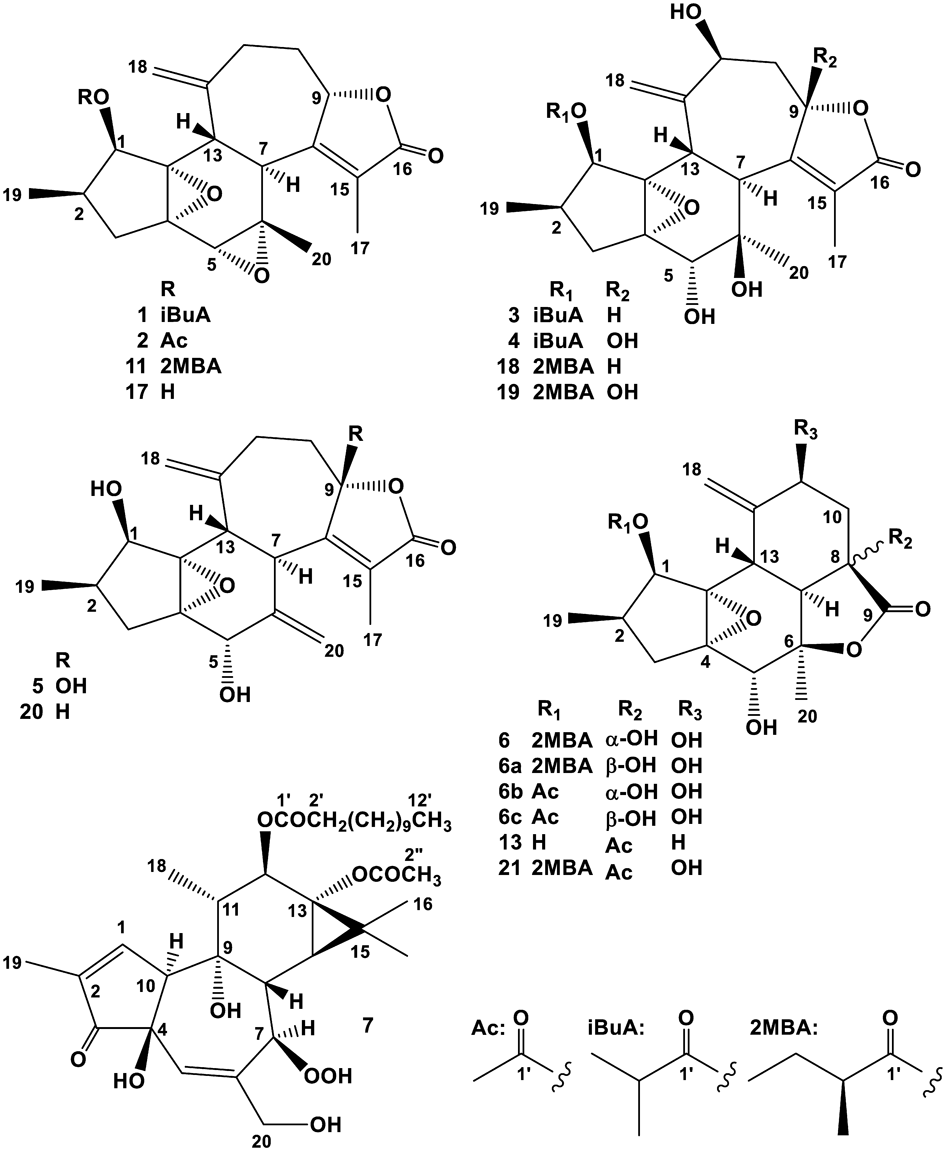
Crotocascarin R (1), [α]26D +13.1, was isolated as an amorphous powder, and its elemental composition was determined to be C24H30O6 through the observation of a quasi-molecular ion peak [M + Na]+ in high-resolution (HR)–electrospray ionization (ESI)-MS. The IR spectrum exhibited an absorption band at 1735 cm−1, assignable to a ketone-stretching vibration in a γ-lactone. The 1H-NMR spectrum showed two singlet methyl (δH 1.07 and 1.91), one doublet methyl (δH 0.91), and two exo-methylene proton [δH 5.07 (br s) and 5.09 (s)] resonances. Judging from two further doublet methyl signals (δH 1.20 and 1.22) together with a septet signal at δH 2.56, an isopropyl functional group was expected to be present in the molecule (Table 1). The 13C-NMR spectroscopic data were essentially superimposable to that of crotocascarin D (11) (Table 2), isolated from the stems of the title plant, except for the acyl region3) (Fig. 1). Therefore, the structure of crotocascarin R (1) was elucidated to be crotocascarin I (17)4) 1-O-isobutyrate, as shown in Fig. 1. The positive and negative Cotton effects at 252 and 221 nm, respectively, which was the same Cotton effects of 11 and 17 suggested that the absolute stereochemistry of them were same.
Table 1.
1H-NMR Spectroscopic Data for Crotocascarins R–V (
1–
5), Crotocascarin δ (
6) (600 MHz, CDCl
3)
| H | 1 | 2 | 3 | 4 | 5 | 6 |
|---|
| 1 | 5.49 d 5 | 5.48 d 5 | 5.41 d 5 | 5.40 d 5 | 4.15 d 5 | 5.79 d 5 |
| 2 | 2.21 m | 2.21 m | 2.13 m | 2.13 m | 2.03 m | 2.21 m |
| 3a | 2.48 dd 14, 7 | 2.48 dd 14, 7 | 2.45 dd 13, 7 | 2.44 dd 14, 7 | 2.40 dd 13, 7 | 2.35 dd 14, 7 |
| 3b | 1.65 dd 14, 11 | 1.65 dd 14, 10 | 1.60 dd 13, 10 | 1.64 dd 14, 10 | 1.53 dd 13, 10 | 1.55 dd 14, 10 |
| 5 | 3.15 s | 3.14 s | 3.61 s | 3.71 s | 4.42 s | 4.38 s |
| 7 | 3.09 d 13 | 3.08 d 13 | 2.81 d 11 | 2.91 d 11 | 3.50 d 12 | 2.20 d 13 |
| 9 | 4.85 m | 4.88 m | 5.22 m | | | |
| 10a | 2.60–2.57 m | 2.60–2.55 m | 2.64 ddd 14, 5, 5 | 2.72 dd 14, 4 | 2.54 ddd 13, 4, 4 | 2.21 dd 15, 6 |
| 10b | 1.17 m | 1.16 m | 1.34 ddd 14, 9, 4 | 1.67 dd 14, 4 | 1.63 m | 2.09 dd 15, 8 |
| 11 | 2.60–2.57 2H m | 2.60–2.55 2H m | 4.34 dd 5, 4 | 4.41 dd 4, 4 | 2.46 2H m | 4.51 dd 8, 6 |
| 13 | 2.57 d 13 | 2.60 d 13 | 3.73 d 11 | 3.96 d 11 | 3.34 d 12 | 3.04 d 13 |
| 17 | 1.91 3H br s | 1.91 3H br s | 1.78 3H d 1 | 1.78 3H br s | 1.69 3H br s | |
| 18a | 5.09 s | 5.08 s | 5.32 s | 5.31 s | 5.22 s | 5.38 s |
| 18b | 5.07 s | 5.07 s | 5.22 s | 5.23 s | 5.09 s | 5.28 s |
| 19 | 0.91 3H d 7 | 0.92 3H d 7 | 0.92 3H d 7 | 0.92 3H d 7 | 1.00 3H d 7 | 0.91 3H d 7 |
| 20 | 1.07 3H s | 1.08 3H s | 1.19 3H s | 1.15 3H s | 5.12 s | 1.59 3H s |
| | | | | 4.76 br s | |
| 2′ | 2.56 m | 2.10 3H s | 2.65 septet 7 | 2.62 septet 7 | | 2.34 qt 7, 7 |
| 3′ | 1.22 3H d 7 | | 1.22 3H d 7 | 1.20 3H d 7 | | 1.65 dqd 14, 7, 7 |
| | | | | | 1.42 dqd 14, 7, 7 |
| 4′ | 1.20 3H d 7 | | 1.20 3H d 7 | 1.18 3H d 7 | | 0.89 3H t 7 |
| 5′ | | | | | | 1.11 3H d 7 |
m: multiplet or overlapped.
Table 2.
13C-NMR Spectroscopic Data for Crotocascarins R–V (
1–
5), Crotocascarin δ (
6) (150 MHz, CDCl
3), and Reference Compounds (
11,
18,
19,
20, and
21) (100 MHz, CDCl
3)
| C | 1 | 11a) | 2 | 3 | 18b) | 4 | 19b) | 5 | 20c) | 6 | 21a) |
|---|
| 1 | 74.4 | 74.5 | 74.9 | 74.2 | 75.1 | 75.4 | 75.4 | 73.22 | 73.1 | 75.3 | 75.5 |
| 2 | 33.6 | 33.2 | 33.5 | 32.4 | 32.4 | 32.4 | 32.8 | 33.2 | 33.2 | 34.6 | 34.4 |
| 3 | 36.9 | 36.9 | 36.8 | 37.3 | 37.3 | 37.2 | 37.0 | 35.8 | 35.8 | 34.4 | 34.3 |
| 4 | 60.5 | 60.5 | 60.5 | 66.4 | 66.4 | 66.7 | 66.5 | 67.5 | 67.6 | 64.7 | 64.8 |
| 5 | 57.6 | 57.6 | 57.6 | 74.2 | 74.2 | 72.8 | 72.7 | 71.1 | 71.1 | 75.5 | 75.4 |
| 6 | 56.1 | 56.1 | 56.1 | 75.5 | 75.4 | 75.0 | 75.5 | 145.4 | 145.1 | 88.8 | 88.0 |
| 7 | 44.0 | 44.0 | 43.9 | 41.6 | 41.7 | 42.0 | 41.9 | 39.7 | 39.5 | 54.1 | 48.0 |
| 8 | 161.6 | 161.7 | 161.7 | 162.8 | 162.9 | 158.2 | 158.8 | 159.7 | 163.1 | 75.1 | 60.1 |
| 9 | 82.2 | 82.2 | 82.2 | 79.8 | 79.9 | 106.9 | 106.6 | 107.7 | 82.6 | 176.5 | 174.7 |
| 10 | 37.7 | 37.7 | 37.7 | 44.6 | 44.4 | 48.4 | 47.7 | 40.9 | 37.2 | 40.5 | 35.8 |
| 11 | 36.2 | 36.2 | 36.3 | 72.6 | 72.5 | 73.4 | 73.6 | 35.4 | 37.0 | 68.2 | 67.9 |
| 12 | 146.0 | 146.0 | 146.0 | 148.6 | 148.6 | 147.8 | 147.5 | 147.5 | 147.2 | 146.6 | 146.1 |
| 13 | 40.7 | 40.7 | 40.7 | 29.3 | 29.5 | 29.4 | 29.3 | 41.7 | 43.3 | 32.5 | 32.4 |
| 14 | 68.8 | 68.7 | 68.7 | 71.8 | 71.7 | 71.5 | 71.2 | 73.18 | 73.0 | 66.4 | 66.0 |
| 15 | 128.6 | 128.5 | 128.6 | 127.0 | 127.0 | 130.3 | 130.2 | 129.4 | 127.2 | | 202.0 |
| 16 | 173.0 | 173.0 | 173.0 | 173.8 | 174.0 | 171.0 | 170.1 | 171.0 | 173.6 | | |
| 17 | 9.6 | 9.6 | 9.6 | 11.2 | 11.2 | 11.5 | 11.2 | 9.3 | 9.3 | | 26.0 |
| 18 | 115.2 | 115.2 | 115.1 | 115.9 | 116.0 | 116.2 | 116.5 | 114.3 | 114.3 | 113.0 | 113.0 |
| 19 | 12.5 | 12.7 | 12.5 | 12.2 | 12.3 | 12.2 | 12.3 | 12.3 | 12.2 | 12.6 | 12.6 |
| 20 | 19.5 | 19.5 | 19.6 | 24.3 | 24.2 | 24.4 | 24.4 | 115.0 | 114.1 | 22.6 | 22.0 |
| 1′ | 175.6 | 175.2 | 169.4 | 178.2 | 178.0 | 178.5 | 178.9 | | | 175.5 | 175.5 |
| 2′ | 34.1 | 41.2 | 20.6 | 34.6 | 41.4 | 34.6 | 41.5 | | | 41.2 | 41.2 |
| 3′ | 19.3 | 26.8 | | 19.1 | 26.2 | 19.0 | 26.2 | | | 26.6 | 26.6 |
| 4′ | 19.5 | 11.7 | | 18.8 | 11.6 | 18.9 | 11.6 | | | 11.8 | 11.8 |
| 5′ | | 17.1 | | | 16.5 | | 16.7 | | | 16.9 | 16.9 |
a) Data from Ref. 3. b) Data from Ref. 5. c) Data from Ref. 4.
Crotocascarin S (2), [α]25D −3.2, was isolated as an amorphous powder, and its elemental composition was determined to be C22H26O6 through the observation of a quasi-molecular ion peak [M + Na]+ in HR-ESI-MS. The IR spectrum also exhibited an absorption band assignable to a ketonic functional group (1752 cm−1) in a γ-lactone. The NMR spectroscopic data of 2 were essentially the same as that of crotocascarin R (1) (Table 2), with the isobutyryl group found in 1 being replaced by an acetyl group [δH 2.10 (3H, s) and δC 20.6 and 169.4]. Therefore, the structure of 2 was elucidated to be crotocascarin I (17) 1-O-acetate, as shown in Fig. 1, and the possibility of introduction of the acetyl functional group during the isolation procedure could not be excluded.
Crotocascarin T (3), [α]20D +105.3, was isolated as an amorphous powder, and its elemental composition was determined to be C24H32O8. The presence of a γ-lactone was deduced by an absorption band at 1736 cm−1 in the IR spectrum, as observed in 1 and 2. Signals assignable to the isopropyl group [δH 1.20 (3H, d, J = 7 Hz, H3-4′), 1.22 (3H, d, J = 7 Hz, H3-3′), and 2.65 (1H, septet, J = 7 Hz)] were observed in the 1H-NMR spectrum, and the remaining 1H- and 13C-NMR signals were essentially the same as those of crotocascarin P,5) which had (S)-(+)-2-methylbutyric acid as an acyl moiety. Therefore, the acyl moiety in crotocascarin P (18)5) (Fig. 1) was expected to be replaced by the isobutyric acid to constitute crotoccascarin T (3), and the structure of 3 is as shown in Fig. 1. Compound 3 showed a significant positive Cotton effect at 219 nm, similar to that of crotocascarin P (18).5)
Crotocascarin U (4), [α]20D +81.4, was isolated as an amorphous powder, and its elemental composition was determined to be C24H32O9, which was one more oxygen atom than that of 3. In the 1H-NMR spectrum, the H-9 signal observed in 3 disappeared, while C-9 (δC 79.8), with a hydrogen atom in 3, was shifted downfield to δC 106.9 by the formation of a hemiketal carbon. The NMR spectra were essentially the same as those of crotocascarin O (19)5) (Fig. 1) except for the acyl region. The positive and negative Cotton effects at 255 and 229 nm, respectively, were similar to those of 19. Therefore, the acyl moiety in crotocascarin O was replaced by isobutyric acid to establish the overall structure of crotoccascarin U (4), as shown in Fig. 1.
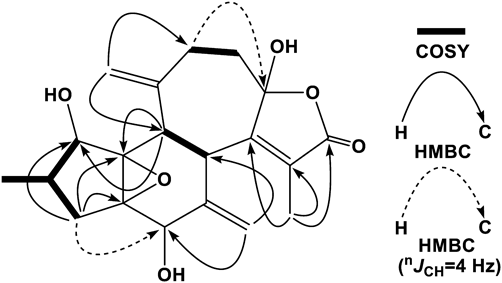
Crotocascarin V (5), [α]26D +105.6, was isolated as an amorphous powder, and its elemental composition was determined to be C20H24O6. The IR absorption band for a γ-lactone was also observed, and similar to crotocascarin K (20), the 1H-NMR and HSQC spectroscopic data of 5 indicated the presence of two exo-cyclic methylenes [δH 5.09 (1H, s) and 5.22 (1H, s) on δC 114.3 (t) and δH 4.76 (1H, br s) and 5.12 (1H, s) on δC 115.0 (t)]. The 13C-NMR data for the five- and six-membered ring regions (C-1 to C-7, C-13, and C-14) were superimposable to that of crotocascarin K (20), and that of the γ-lactone moiety showed close resemblance to that of crotocascarin U (4) (Table 2). Therefore, the oxymethine carbon (C-9) (δC 82.6) in crotocascarin K was expected to be replaced by the hemiketal (δC 107.7) functional group. In the HMBC spectra, correlations (nJCH = 8 Hz) from the exo-methylene protons (δH 5.09 and 5.22, H2-18) to C-11 (δC 35.4) and then correlation (nJCH = 4 Hz) from δH 2.46 (2H, H2-11) to δC 107.7 (C-9) were observed. COSY spectrum together with a significant HMBC correlation from H-13 (δH 3.34) to C-1 (δC 73.22) and/or C-14 (δC 73.18) (nJCH = 8 Hz), and an apparent one from H-3a (δH 2.40) to C-5 (δC 71.1) (nJCH = 4 Hz) (Fig. 2) suggested the structure of 5, as shown in Fig. 1. The similar positive and negative Cotton effects at 249 and 226 nm, respectively, in the electronic circular dichroism (ECD) spectrum to those of crotocascarin O (19) indicated that both have the same absolute stereochemistry.
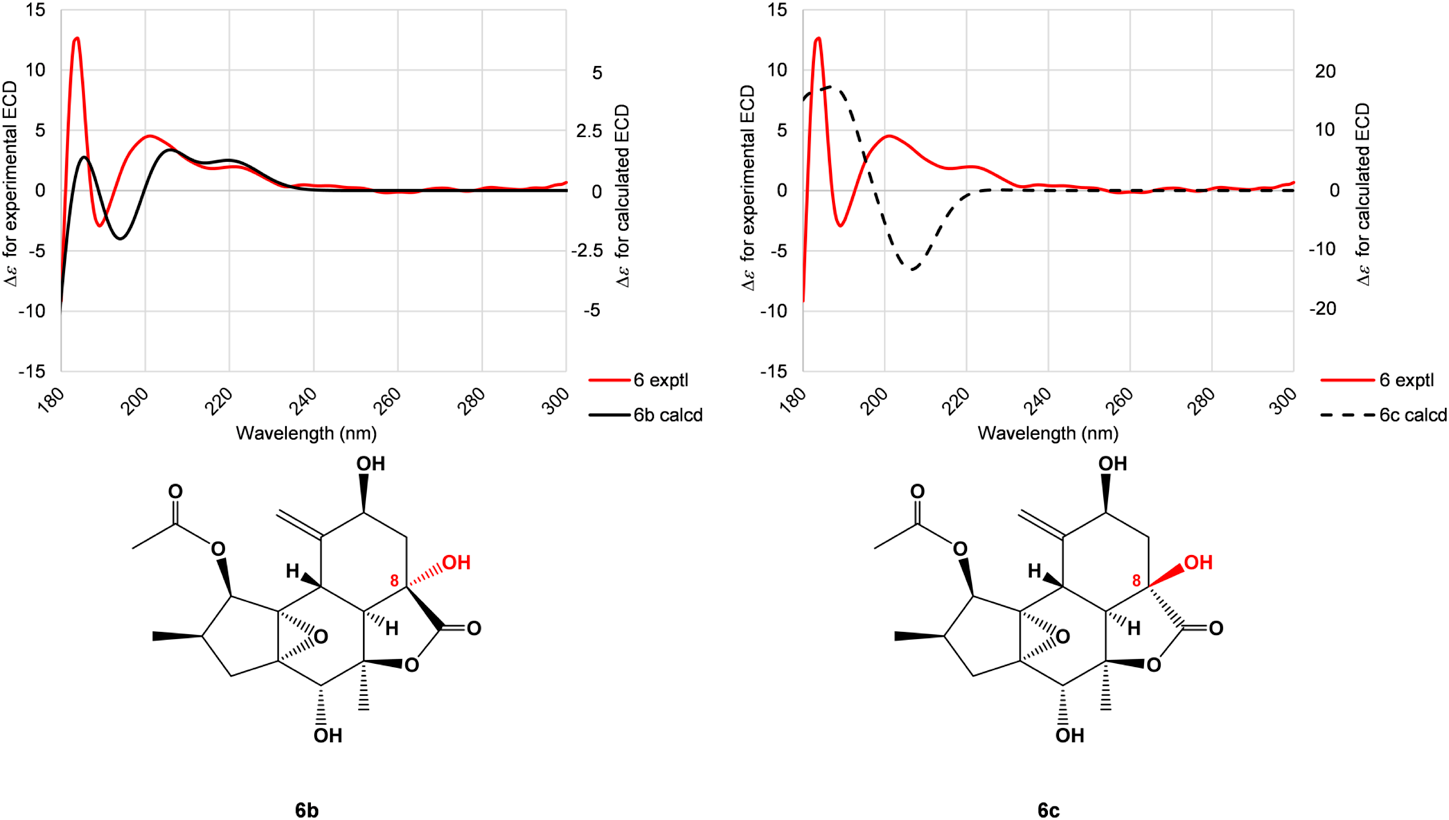
Crotocascarin δ (6), [α]25D +4.7, was isolated as an amorphous powder, and its elemental composition was determined to be C22H30O8. The IR spectrum showed absorption bands ascribable to a saturated γ-lactone (1762 cm−1) and an ester group (1748 cm−1). In the 1H-NMR spectrum, one triplet, two doublet, and one singlet methyls, together with exo-methylene [δH 5.38 (s) and 5.28 (s)] protons, were observed. Proton connectivity from H3-4′ through H3-5′ through H2-3′ and H-2′, observed in the COSY spectrum, evidenced the presence of 2-methybutyric acid moiety (Fig. 3); thus, the core part of this compound was composed of 17 carbons. The 13C-NMR signals of the core part actually comprised those of two methyls, two methylenes, and six methines, three of which bore an oxygen atom, four oxygenated tertiary carbons, one carbonyl carbon, and an exo-cyclic double bond. A closely related analogue, crotocascarin γ (13),4) was isolated in this experiment and further structurally resembled the compound crotocascarin γ 1-O-2-methylbutyrate, known as crotocascarin α (21).3) When comparing the 13C-NMR chemical shifts of 6 and 21, those of C-7, C-8, C-9, and C-10 showed significant discrepancy (Table 2), and two carbon signals for the acetyl group (δC 26.0 and 202.0) in 21 were missing in 6. The elemental composition assumed that the missing acetyl group must be replaced by a hydroxy group, and this assumption was supported by HMBC correlations between H-7 (δH 2.20) and H2-10 (δH 2.09 and 2.21) and C-8 (δC 75.1) (Fig. 3). Thus, the structure of 6 was elucidated to be a deacetyl and hydroxylated compound of 21 at the 8-position, which could be considered as a rearranged trinorcrotofolane. Figure 1 shows two possible isomers (6 and 6a). The orientation of the hydroxy group at the 8-position left some ambiguity as a result of NMR chemical shift simulation using the ChemDraw software, which showed that the chemical shifts of both isomers were quite close, and an attempt at recrystallization for a suitable crystal for X-ray crystallography was not successful. Therefore, the configuration at the 8-position was deduced by the comparison of experimental and theoretical ECD spectra12) (Fig. 4). Figure 4 shows two calculated ECD spectra for 6b and 6c (solid and dotted lines in black, respectively) as well as the actual experimental spectrum (6) (solid line in red). The comparisons of these spectra indicated that crotocascarin δ (6) possessed the hydroxy group at the 8-position in the α-face, namely, the 8S configuration. An HPLC analysis of 2-methybutyric acid moiety and a commercially available authentic material using a chiral detector revealed that the acyl moiety had an S configuration. Finally, the structure of crotocascarin δ (6) was elucidated to be that shown in Fig. 1.
12-O-Lauroyl-7-peroxy-5-ene-phorbol-13-acetate (7), [α]25D +46.8, was isolated as an amorphous powder, and its elemental composition was determined to be C34H52O10. The IR spectrum showed absorption bands for the ketonic functional groups (1716 and 1730 cm−1) and the UV spectrum for an α,β-unsaturated ketone (227 nm). The superposed peaks observed in the 1H-NMR at δH 1.20–1.31 and in the 13C-NMR at δC 29.1, 29.30, 29.32, 29.5, and 29.6 × 2 (Table 3), together with a typical triplet signal [δH 0.88 (3H, t, J = 7 Hz) on δC 14.1], indicated the presence of a fatty acid. An acetyl group [δH 2.10 (3H, s) and δC 21.1 and 174.1] was also found in a part of compound 7; thus, the core unit was made of 20 carbons. A closely related compound, tigliane-type diterpene (a phorbol derivative) (22), whose NMR data were essentially superimposable to that of 7, was isolated from Sapium sebiferum as an Epstein–Barr virus–inducing activity13) (Table 3). Compound 7 was positive to the Griess reagent for a hydroperoxide, and the mass spectral data required that only one fatty acid species, lauric acid (C-12), be present in 7 as a medium-chain acyl moiety. Ohigashi et al. also claimed that only one molecular species of a fatty acid, palmitic acid, was present in the phorbol hydroperoxide derivative.13) Therefore, the structure of 7 contained lauric acid, which is expected to be a new compound, as shown in Fig. 1.
Table 3.
13C-NMR Spectroscopic Data for Phorbol Derivatives (
7 and
22) (100 MHz, CDCl
3)
| C | 7 | | 22a) |
|---|
| 1 | 160.2 | | 160.1 |
| 2 | 134.1 | | 134.1 |
| 3 | 206.5 | | 206.6 |
| 4 | 72.5 | | 72.5 |
| 5 | 134.1 | | 133.5 |
| 6 | 150.7 | | 150.8 |
| 7 | 83.1 | | 83.0 |
| 8 | 46.0 | | 45.8 |
| 9 | 75.0 | | 75.2 |
| 10 | 57.3 | | 57.2 |
| 11 | 44.3 | | 44.2 |
| 12 | 76.7 | | 76.6 |
| 13 | 66.1 | | 66.1 |
| 14 | 31.5 | | 31.4 |
| 15 | 25.8 | | 25.8 |
| 16 | 23.8 | | 23.8 |
| 17 | 16.6 | | 16.6 |
| 18 | 14.7 | | 14.7 |
| 19 | 10.2 | | 10.3 |
| 20 | 66.4 | | 66.1 |
| 1′ | 173.8 | | 173.9 |
| 2′ | 34.6 | | 34.6 |
| 3′ | 31.9 | | 31.9 |
| 29.1 | | 29.0 |
| 29.30 | | 29.3 |
| 4′–9′ | 29.32 | 4′–13′b) | 29.4 |
| 29.5 | | 29.6 |
| 29.6 × 2 | | 29.7 |
| 10′ | 25.8 | 14′ | 25.2 |
| 11′ | 22.7 | 15′ | 22.7 |
| 12′ | 14.1 | 16′ | 14.1 |
| 1″ | 174.1 | | 174.1 |
| 2″ | 21.1 | | 21.1 |
a) Data from Ref. 13 (at 50 MHz). b) Some are double or triple strengths.
Experimental
General Experimental ProceduresThe optical rotations were measured on a JASCO P-1030 digital polarimeter. The IR and UV spectra were measured on Horiba FT-710 and JASCO V-520 or V-670 UV/Vis spectrophotometers, respectively. The 1H- and 13C-NMR spectra were taken on a JEOL JNM α-400 spectrometer at 400 and 100 MHz and a Bruker Avance III spectrometer at 600 and 150 MHz, respectively, with tetramethylsilane as an internal standard. The ECD spectra were obtained with the JASCO J-720 and J-1100 spectropolarimeters. Positive-ion HR-ESI-MS was performed with an Applied Biosystems QSTAR® XL NanoSpray™ System.
Silica gel CC was performed on silica gel 60 (E. Merck, Darmstadt, Germany) and ODS open CC on Cosmosil 75C18-OPN (Nacalai Tesque, Kyoto, Japan) [Φ = 50 mm, L = 25 cm, linear gradient: MeOH–H2O (1 : 1, 1 L)→MeOH (1 L), MeOH (1 L)→CHCl3–MeOH (1 : 1, 1 L), fractions of 10 g being collected]. HPLC was performed on an octadecyl silica (ODS) column (Inertsil; GL Science, Tokyo, Japan; Φ = 6.0 mm, L = 250 mm, flow rate: 1.6 mL/min), and the eluate was monitored with a refractive index monitor. (S)-(+)-2-Methylbutyric acid was purchased from Sigma-Aldrich Corp. (St. Louis, MO, U.S.A.).
Plant MaterialThe leaves of C. cascarilloides Räuschel (Euphorbiaceae) were collected in Kunigami-son, Kunigami-gun, Okinawa, Japan, in July 2004, and a voucher specimen was deposited in the Herbarium of Pharmaceutical Sciences, Graduate School of Biomedical and Health Sciences, Hiroshima University (04-CC-Okinawa-0628). The plant was identified by one of the authors (TS).
Extraction and IsolationThe extraction and fractionation procedures were previously described; that is, the material (6.53 kg) yielded dry extracts to give 59.1 g, 100 g, 126 g and 263 g of n-hexane, EtOAc, 1-BuOH and H2O-soluble fractions, respectively.2) The EtOAc-soluble fraction was subjected to silica gel CC (Φ = 80 mm, L = 40 cm) and eluted with n-hexane (6 L), n-hexane–EtOAc [(9 : 1, 6 L), (17 : 3, 6 L), (4 : 1, 6 L), (3 : 1, 6 L), (7 : 3, 6 L), (13 : 7, 6 L), (3 : 2, 6 L), (1 : 1, 6 L), (9 : 11, 6 L), (2 : 3, 6 L), and (3 : 7, 6 L)], EtOAc (6 L), acetone (6 L), and MeOH (6 L), with 2 L fractions being collected. The residue (3.56 g) in fractions 26–27 was separated via ODS open CC to give fr. 2 (379.4 mg) in fractions 12–16, fr. 3 (517 mg) in fractions 21–43, fr. 7 (404 mg) in fractions 66–95, and fr. 10 (154 mg) in fractions 158–172. fr. 2 was again subjected to silica gel CC [Φ = 10 mm, L = 40 cm, linear gradient: CHCl3 (500 mL)→CHCl3–MeOH (19 : 1, 500 mL), with 5 g fractions being collected] to give the residues (35.0 mg) in fractions 88–96 and (29.3 mg) in fractions 106–121. The former was separated via HPLC (H2O–MeOH, 1 : 1) to afford 5.4 mg of 3 from the peak at 20.2 min and 6.0 mg of 4 from the peak at 21.6 min. The latter was also purified via HPLC (H2O–MeOH, 1 : 1) to give 5.7 mg of 13 from the peak at 12.0 min. fr. 3 was subjected to silica gel CC [Φ = 10 mm, L = 40 cm, linear gradient: CHCl3 (500 mL)→CHCl3–MeOH (19 : 1, 500 mL), with 5 g fractions being collected] to give the residues (33.2 mg) in fractions 28–31, (11.6 mg) in fractions 134–153, and (10.0 mg) in fractions 154–170. The first was purified via HPLC (H2O–MeOH, 2 : 3) to yield 13.5 mg of 2 from the peak at 16.2 min. The second was purified via HPLC (H2O–MeOH, 2 : 3) to afford 2.9 mg of 6 from the peak at 8.5 min. The third was purified via HPLC (H2O–MeOH, 2 : 3) to afford 3.5 mg of 5 from the peak at 5.5 min. fr. 7 was subjected to silica gel CC [Φ = 10 mm, L = 40 cm, linear gradient: n-hexane (500 mL)→EtOAc (500 mL), with fractions of 5 g being collected] to give 70.6 mg of 10 in fractions 56–60 and three residues (21.7 mg) in fractions 43–50, (41.1 mg) in fractions 51–55, and (56.3 mg) in fractions 61–68. The first residue was purified via HPLC (H2O–MeOH, 7 : 13) to afford 1.6 mg of 1, 2.7 mg of 8, and 3.5 mg of 11 from the peaks at 21.5 min, 30.9 min, and 33.2 min, respectively. The second residue was purified via silica gel CC [Φ = 10 mm, L = 40 cm, CHCl3 (100 mL), CHCl3–acetone (99 : 1 and 49 : 1, 100 mL each), and CHCl3–MeOH (1 : 1, 100 mL), with fractions of 5 g being collected] to afford 19.7 mg of 9 in fractions 40–45. The third residue was purified via silica gel CC [Φ = 10 mm, L = 40 cm, linear gradient: CHCl3 (500 mL)→CHCl3–MeOH (19 : 1, 500 mL), with fractions of 5 g being collected] to afford 7.4 mg of 12 in fractions 55–63. fr. 10 was subjected to silica gel CC [Φ = 10 mm, L = 40 cm, linear gradient: n-hexane (500 mL)→EtOAc (500 mL), with fractions of 5 g being collected] to give 16 (6.3 mg) in fraction 66. The residue (29.8 mg) in fractions 68–69 was purified via HPLC (H2O–MeOH, 3 : 17) to afford 8.4 mg of 7 from the peak at 18.5 min.
The residue (4.04 g) in fractions 28–30 obtained on the first silica gel CC was separated via ODS open CC to give 83.1 mg of 14 and 94.8 mg of 15 in fractions 146–152 and 164–170, respectively.
Crotocascarin R (1)White amorphous powder, [α]26D +13.1 (c = 0.11, CHCl3); IR νmax (film) cm−1: 2964, 2924, 2875, 1735, 1654, 1457, 1328, 1185, 1139, 977; UV λmax (MeOH) nm (log ε): 249 sh (3.17), 216 (3.99); 1H-NMR (600 MHz, CDCl3): Table 1; 13C-NMR (150 MHz, CDCl3): Table 2; ECD ∆ε (nm): +0.85 (252), −2.11 (221) (c = 2.31 × 10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 437.1928 [M + Na]+ (Calcd for C24H30O6Na: 437.1934).
Crotocascarin S (2)Off-white amorphous powder, [α]25D −3.2 (c = 0.97, CHCl3); IR νmax (KBr) cm−1: 2971, 292 5, 2879, 1752, 1737, 1651, 1456, 1336, 1165, 984; UV λmax (MeOH) nm (log ε): 250 sh (2.88), 215 (3.97); 1H-NMR (600 MHz, CDCl3): Table 1; 13C-NMR (150 MHz, CDCl3): Table 2; ECD ∆ε (nm): +0.79 (251), −4.71 (210) (c = 2.50 × 10−5 M); HR-ESI-MS (positive-ion mode) m/z: 409.1616 [M + Na]+ (Calcd for C22H26O6Na: 409.1621).
Crotocascarin T (3)White amorphous powder, [α]20D +105.3 (c = 0.40 CHCl3); IR νmax (film) cm−1: 3480, 2974, 2934, 1736, 1654, 1457, 1336, 1205, 1151, 922; UV λmax (MeOH) nm (log ε): 256 sh (2.80), 225 (3.93); 1H-NMR (600 MHz, CDCl3): Table 1; 13C-NMR (150 MHz, CDCl3): Table 2; ECD ∆ε (nm): +2.96 (219) (c = 4.47 × 10−5 M); HR-ESI-MS (positive-ion mode) m/z: 471.2002 [M + Na]+ (Calcd for C24H32O8Na: 471.1989).
Crotocascarin U (4)White amorphous powder, [α]20D +81.4 (c = 0.36 CHCl3); IR νmax (film) cm−1: 3500, 2975, 2933, 1738, 1652, 1458, 1333, 1212, 1151, 929; UV λmax (MeOH) nm (log ε): 257 sh (2.92), 220 (3.80); 1H-NMR (600 MHz, CDCl3): Table 1; 13C-NMR (150 MHz, CDCl3): Table 2; ECD ∆ε (nm): +4.59 (255), −8.76 (229) (c = 3.88 × 10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 487.1932 [M + Na]+ (Calcd for C24H32O9Na: 487.1938).
Crotocascarin V (5)Off-white amorphous powder, [α]26D +105.6 (c = 0.19, CHCl3); IR νmax (film) cm−1: 3490, 2949, 2932, 2879, 1748, 1646, 1457, 1265, 1191; UV λmax (MeOH) nm (log ε): 244 sh (2.38), 212 (3.77); 1H-NMR (600 MHz, CDCl3): Table 1; 13C-NMR (150 MHz, CDCl3): Table 2; ECD ∆ε (nm): +4.77 (249), −1.28 (226) (c = 2.59 × 10−5 M, MeOH); HR-ESI-MS (positive-ion mode) m/z: 383.1472 [M + Na]+ (Calcd for C20H24O6Na: 383.1465).
Crotocascarin δ (6)White amorphous powder; [α]25D +4.7 (c = 0.13, CHCl3); IR νmax (KBr) cm−1: 3435, 2971, 2931, 2880, 1762, 1748, 1653, 1457, 1136; UV λmax (CH3CN) nm (log ε): 220 sh (3.31); 1H-NMR (600 MHz, CDCl3): Table 1; 13C-NMR (150 MHz, CDCl3): Table 2; ECD ∆ε (nm): +12.6 (184), −2.91 (189), +4.52 (201), +1.98 (221) (c = 4.74 × 10−5 M, CH3CN); HR-ESI-MS (positive-ion mode) m/z: 445.1830 [M + Na]+ (Calcd for C22H30O8Na: 445.1832).
12-O-Lauroyl-7-peroxy-5-ene-phorbol-13-acetate (7)Off-white amorphous powder, [α]25D +46.8 (c = 0.31, CHCl3); IR νmax (film) cm−1: 3392, 2954, 2926, 1730, 1716, 1628, 1458, 1265, 1161; UV λmax (MeOH) nm (log ε): 227 (3.85); 1H-NMR (400 MHz, CDCl3) δ: 10.51 (1H, s, -OOH), 7.63 (1H, br s, H-1), 6.38 (1H, d, J = 2 Hz, H-5), 5.46 (1H, d, J = 10 Hz, H-12), 4.76 (1H, dd, J = 9, 2 Hz, H-7), 4.45 (1H, d, J = 11 Hz, H-20a), 4.23 (1H, d, J = 11 Hz, H-20b), 2.98 (1H, dd, J = 3, 2 Hz, H-10), 2.85 (1H, dd, J = 9, 6 Hz, H-8), 2.33 (2H, t, J = 7 Hz, H2-2′), 2.19 (1H, dd, J = 10, 6 Hz, H-11), 2.10 (3H, s, H3-2″), 1.79 (3H, d, J = 2 Hz, H3-19), 1.66 (1H, d, J = 6 Hz, H-14), 1.31–1.20 (18H, m, -CH2- × 9), 1.28 (3H, s, H3-16), 1.22 (3H, s, H3-17), 0.92 (3H, d, J = 6 Hz, H3-18), 0.88 (3H, t, J = 7 Hz, H3-12′); 13C-NMR (100 MHz, CDCl3): Table 3; HR-ESI-MS (positive-ion mode) m/z: 643.3446 [M + Na]+ (Calcd for C34H52O10Na: 643.3452).
Theoretical ECD Calculation of 6b and 6c for Absolute Configuration at Position 8 of Crotocascarin δ (6)Virtual 1-O-acetyl derivatives (6b and 6c), instead of natural (S)-(+)2-methylbutyric acid esters (6 and 6a), were used for calculation to minimize conformational complexities. Conformational analyses for 6b and 6c were performed with the Spartan′20 V1.0.0. program (Wavefunction Inc. Irvine, CA, U.S.A.) on a commercially available personal computer (operating system: Microsoft 64-bit version of Windows 10 Home edition; quad-core central processing unit: Core i7-8550U processor (Intel Corp., Santa Clara, CA, U.S.A.) run at 1.80 GHz; random access memory: 8 GB). Stable conformers up to 40 kcal/mol for 6b and 6c were initially searched using the Merck molecular force field method. Then the stable conformers suggested were further optimized using the Hartree–Fock (HF)/3-21G and ωB97XD/6-31G* programs. The resulting conformers were subjected to ECD calculation, and the ECD calculations for these conformers were performed with Gaussian16 (Revision A.03 by Gaussian)14) on the ChemPark Cloud system.15) The dominant conformers of 6b and 6c capable of covering >90% of the population according to Boltzmann distribution were selected. Time-dependent density functional theory calculations were conducted at the PBE0/TZVP level for these conformers. The resulting rotational strength data were converted to Gaussian curves (bandwidth sigma = 0.45 eV) to obtain the ECD spectra of each conformer, and the spectra were combined after Boltzmann weighting according to their population contributions. The wavelength of the spectra was corrected (−5 nm) based on the absorptions of about 220 nm (referring to the experimental and calculated UV spectra) to give the corresponding theoretical ECD spectra.
HPLC Analysis of (S)-(+)-2-Methylbutyric Acid in Crotocascarin δ (6)Crotocascarin δ (6) (0.5 mg) and authentic (S)-(+)-2-methylbutyric acid (500 µL) were each dissolved in 1 mL of a 1 : 1 mixture of 10% KOH in H2O and 50% aqueous 1,4-dioxane and then heated for 3 h at 100 °C. The cooled reaction mixtures were acidified with one drop of conc. HCl, and then the filtrates were evaporated. The two residues were analyzed via HPLC (column: Inertsil ODS-3, 6 × 250 mm; solvent: 20% acetonitrile in H2O containing 0.5% phosphoric acid; flow rate: 1.6 mL/min) with a chiral detector (JASCO OR-4090), which showed a peak of (S)-(+)-2-methylbutyric acid at 16.8 min with a positive optical rotation sign.