Regular Articles
Model Synthetic Study of Tutin, a Picrotoxane-Type Sesquiterpene: Stereoselective Construction of a cis-Fused 5,6-Ring Skeleton
2022 Volume 70 Issue 6 Pages 435-442
Details
2022 Volume 70 Issue 6 Pages 435-442
Picrotoxinin, coriamyrtin, and tutin are representative natural products classified as picrotoxane-type sesquiterpenes and they function as strong neurotoxins. Because they possess a cis-fused 5,6-ring skeleton with a highly congested functionalization, organic chemistry researchers have pursued the development of a stereoselective synthesis method for such skeleton. This study aims to stereoselectively synthesize the cis-fused 5,6-ring skeleton with two tetrasubstituted carbons at both angular positions using a model compound. The results revealed that the desymmetrization of the 2-methyl-1,3-cyclopentanedione moiety via the DL-proline-mediated intramolecular aldol reaction of a pentanal derivative bearing an isopropenyl group and the five-membered ring at the 3- and 5-position, respectively, provided the desired cis-fused skeleton. This reaction can construct four contiguous stereogenic centers of the bicyclic skeleton with the two angular positions in good yield with high stereoselectivity. Further, this reaction was applied to the kinetic resolution of the racemate using L-proline, providing the enantiomeric pure aldol product with the desired skeleton. This method can be utilized for total synthesis of picrotoxane-type sesquiterpenes.
Picrotoxane-type sesquiterpenes have been found in various plants since the 1900s,1) and the novel natural products belonging to this family have been isolated in the recent years.2–5) Among these, picrotoxinin (1), coriamyrtin (2), and tutin (3) are well-known natural products because they function as strong neurotoxins,6) and are hence used in biological chemistry (Fig. 1a). From an organic chemistry viewpoint, 1–3 have attractive structures with a highly functionalized cis-fused 5,6-ring skeleton with two tetrasubstituted angular carbons. This structure has inspired organic chemistry researchers to develop efficient synthesis methods for these natural products. In fact, the total synthesis of 1 was reported by Shenvi,7) Trost,8) Yoshikoshi,9) Yamada,10) and Corey groups.11) By contrast, there are only two reports on the synthesis of coriamyrtin (2) in the 1980s.10,12) Moreover, as tutin (3) bears the cis-fused skeleton with all carbons functionalized, its total synthesis has only been achieved by the Yamada group,13) albeit it has the disadvantage of requiring a total of 41 steps from an α-tetralone derivative.14) Recently, there are reports on synthetic studies of picrotoxane-type skeleton (4), which lacks the hydroxy group in an angular position.15–17) However, the applications of these methods toward the synthesis of 2 and 3 has not been explored. These backgrounds encouraged us to examine the total synthesis of 3, a method of which can also be developed for the synthesis of 1 and 2. Herein, we describe the stereoselective synthesis of the cis-fused 5,6-ring skeleton with the two tetrasubstituted angular carbons.

A major concern in the total synthesis of tutin (3) is the stereoselective formation of the two stereocenters in the two angular positions of the picrotoxane-type skeleton (4). The introduction of a one-carbon unit at the C-9 position of 4 could be achieved using a carbonyl compound. Thus, we chose enone 5 as the synthetic precursor of 3 (Fig. 1b). This study aimed to synthesize its model compound 6, wherein the two oxy-functionalized groups at the 2- and 3-positions were removed. Because 6 possesses a β-hydroxy carbonyl moiety, we used the intramolecular aldol reaction for the synthesis, thereby retro-synthesizing 6 to 7 with a symmetrical 2-methyl-1,3-cyclopentenedione structure. The desymmetric strategy is a powerful tool for synthesizing a complex molecule with multiple stereogenic centers in organic synthesis.18,19) In fact, reactions for desymmetrization of the 1,3-cyclopentanedione moiety have been reported by various groups.20–27) However, their application in the synthesis of picrotoxane-type sesquiterpenes has not been explored. In this reaction, there are two possible reaction pathways starting from enolate intermediate 8: an intramolecular attack of the enolate anion on carbon A to produce 9 or its attack on carbon B to produce 6 (Fig. 1c). The latter reaction can deliver the desired stereochemistries at the two angular positions. Although the synthesis of 6 requires strict reaction conditions to allow the accurate recognition of the two carbonyl groups, this reaction could also cause synchronous construction of the trans configuration between the carbonyl group and the isopropenyl group in 6 because the intramolecular reaction must occur avoiding their mutual steric repulsion. Thus, this desymmetric strategy could allow the formation of the four contiguous stereogenic centers in a highly stereoselective manner.
Based on the synthetic plan of 6, the intramolecular aldol reaction of ester 10, which was prepared from 2-methyl-1,3-cyclopentanedione (11), was investigated (Chart 1). The Michael reaction of 11 to acrolein, followed by the Wittig reaction with ethyl 2-(triphenylphosphoranylidene)propionate gave trisubstituted (E)-unsaturated ester 12 as a single isomer in high yield. After the bis-acetalization of 12 under the Noyori condition,28) diisobutylaluminum hydride (DIBAL) reduction of the resulting ester 13 provided an allylic alcohol 14. The Jonson–Claisen rearrangement29) of 14 was adopted to construct the isopropenyl group in 10. The formation of keteneacetal 15 proceeded smoothly by treatment of 14 with triethyl orthoacetate and pivalic acid (PivOH) at 50 °C. The reaction mixture was then heated at 130 °C to supply γ,δ-unsaturated ester 16. Subsequent acid hydrolysis to remove the bis-acetal moieties was conducted in one-pot, affording diketone 17 in 72% yield. Finally, copper(II) bromide-mediated oxidation of the 1,3-cyclopentanedione moiety30) in the presence of 2-methyl-2-butene delivered the desired compound 10.
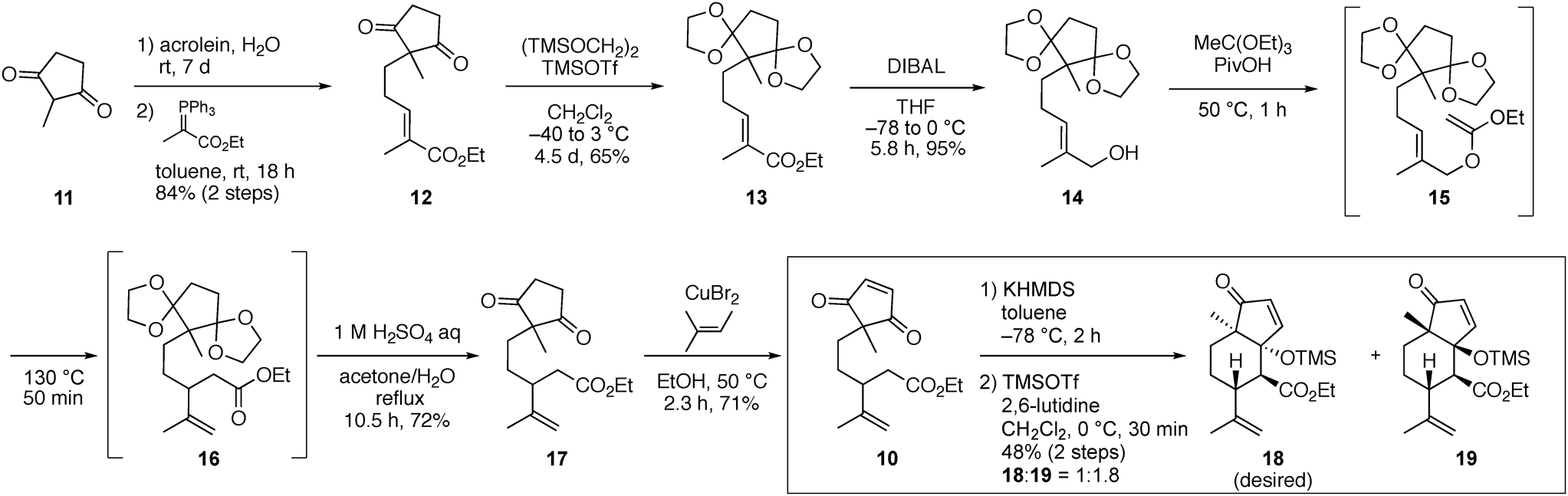
Ph = phenyl, Et = ethyl, Me = methyl.
An investigation of the intramolecular aldol reaction of 10 under various conditions revealed that the use of potassium hexamethyldisilazide (KHMDS) in toluene gave the best result (Chart 1). In this reaction, the counter cation was an important factor because the exchange of potassium to sodium and lithium, respectively, in the base did not provide the aldol products. Further, the use of other bases, such as lithium diisopropylamide, sodium hydride, potassium tert-butoxide, resulted in the decomposition of 10. Although the crude product obtained by exposure of KHMDS in toluene contained the two diastereomeric mixtures, the isolation of each compound in its pure form was difficult owing to the presence of inseparable byproducts. Thus, without purification, trimethylsilyl (TMS)-protection of the hydroxy group of the two desired products in the mixture was conducted using trimethylsilyl triflate (TMSOTf) and 2,6-lutidine, which led to the isolation of two aldol products as a 1.8 : 1 diastereomeric ratio, with in an overall 48% yield for the two-step reaction process.
The NMR analysis of the two products 18 and 19 showed that the desired product, 18, was a minor product (Fig. 2). The JH4–H5 values of 18 and 19 were 12.8 and 12.0 Hz, respectively, which confirmed the trans configurations between the ester and the isopropenyl group in both compounds. In addition, the nuclear Overhauser effect spectroscopy (NOESY) correlation between the proton of the Me group and TMS group at the angular positions was detected in 18 and 19 to ensure the presence of the cis-fused skeleton. By contrast, in the same spectrum, while the H-5 proton in 19 was not correlated to the angular methyl protons, that in 18 was correlated to the corresponding proton. These results showed that 18 bears the desired four contiguous stereogenic centers, and 19 is a diastereomer where the two angular stereogenic centers are opposite to those in 18. Remarkably, the chemical shifts of H-4 and H-5 in 18 and 19 were dramatically different. While the chemical shift value of H-5 was lower than that of H-4 in 18, H-5 was higher than H-4 in 19. This fact would be helpful for the structure confirmation of the cis-fused 5,6-ring skeleton in the picrotoxane-type sesquiterpenes.

Because the intramolecular aldol reaction of 10 mainly gave the undesired cis-fused skeleton 19, we next examined the reaction using other substrates, the synthetic method of which is shown in Chart 2. The Eschenmoser–Claisen rearrangement31) of allylic alcohol 14 in toluene at reflux afforded amide 20 in 76% yield. Reduction of the amide moiety by the Schwartz reagent, prepared from zirconocene dichloride and lithium tri-tert-butoxyaluminum hydride,32) produced aldehyde 21. The bis-acetal structure was removed under acidic hydrolysis conditions, releasing 1,3-cyclopentanedione 22. To oxidize the five-membered ring, 22 was subjected to the same reaction conditions that were used for the conversion of 17 into 10. However, the aldehyde moiety was simultaneously acetalized to produce 23. Thus, the ethyl acetal was converted to the aldehyde in diluted sulfuric acid and tetrahydrofuran (THF) to obtain an aldol precursor 24 with 19% yield in 2 steps. Another precursor 25 was prepared from aldehyde 21 through an oxime formation, followed by acetic anhydride-mediated dehydration of the oxime and removal of the bis-acetal via a one-pot protocol, and then oxidation of the 1,3-cyclopentanedione moiety of the obtained nitrile 26.

Cp = cyclopentadienyl, t-Bu = tertiary-butyl, Ac = acetyl.
The results of the intramolecular aldol reaction using nitrile 25 and aldehyde 24 are shown in Charts 3a and 3b. The reaction of 25 with KHMDS in toluene resulted in the corresponding aldol products 27 and 28, similar to ester 10. However, the total yield and the production ratio of the desired compound 27 dramatically decreased. While the intramolecular aldol reaction of 24 occurred via the use of a weaker base, alumina, the dehydration of the aldol product 29 also proceeded to afford only 30 in 15% yield as a 1.1 : 1 diastereomeric mixture. Although we subjected 24 to various reaction conditions for producing 29 in good yield, it was unsuccessful due to the probable instability of 24 resulting from the cyclopentene-1,3-dione moiety.33) Therefore, we changed the intramolecular aldol reaction reactant 24 to 22 bearing the 1,3-cyclopentanedione moiety (Chart 3c). The treatment of 22 with alumina provided aldol products 31 and 32 in 50% NMR yield, but the major product was 32 with undesired stereochemistries at the two angular positions (condition A). On the other hand, the use of pyrrolidine (condition B) increased the production ratio of the desired product 31, albeit the yield and diastereoselectivity was unsatisfactory (19% isolated yield as a 1 : 2 mixture of 31 and 32). The low yield was attributed to the competitive intermolecular aldol reaction of 22, as compounds derived from the side reaction were detected.

DMSO = dimethyl sulfoxide.
The result obtained using pyrrolidine prompted DL-proline as the choice reagent for the intramolecular aldol reaction of 22, as the intramolecular hydrogen network between one of the carbonyl groups and the enamine intermediate generated in situ will inhibit the intermolecular aldol reaction and increase the production ratio of 31. The reaction proceeded smoothly to provide 31 with high diastereoselectivity, succeeding in its isolation in 73% yield34) (Fig. 3a). The structure of 31 was confirmed by the NMR analysis of benzyl esters 33 and 34. These were prepared from a 1.9 : 1 mixture of 31 and 32 through the Pinnick oxidation, the benzylation of the resulting carboxylic group, and their separation via silica gel chromatography (Fig. 3b). Similar to the NMR results for 18 and 19 in Fig. 2, both JH4–H5 values were more than 12.0 Hz and the chemical shift value of H-5 in 33 was lower than that in 34. In addition, the NOESY spectrum of 33 showed the correlation of H-5 with the Me group and the hydroxy group in the angular positions. This indicates that 31 obtained by the DL-proline mediated intramolecular aldol reaction possessed the same four contiguous stereogenic centers in the cis-fused 5,6-ring skeleton of picrotoxane-type sesquiterpenes.

a The enantiomeric excess (ee) of 31 was evaluated after its transformation to 33. Bn = benzyl. DMF = N,N-dimethylformamide.
This intramolecular aldol reaction was applicable to the asymmetric synthesis of 31 via kinetic resolution (Fig. 3c). Thus, the change of DL-proline to L-proline induced the reaction of either enantiomer of rac-22 preferentially, to provide optically active 31 and diastereomer 32 in 38 and 3% NMR yield, respectively, with the recovery of 20% NMR yield of 22. Because the separation of these three mixtures was difficult, the optical purity of 31 was evaluated after the exposure to the two-step protocol similar to Fig. 3b and the isolation of 33. The value of its enantiomeric excess (≥99% ee) demonstrated that the L-proline mediated intramolecular reaction proceeded via the strong intramolecular hydrogen network. We speculate that the preferred transition state (TS) of the reaction is TS-A, where the absolute configuration of the stereogenic center is R because TS-B derived from (S)-22 results in the steric hindrance between the 1,3-cyclopentanedione moiety and the pyrrolidine moiety (Fig. 3d). There is a similar steric repulsion in TS-C to produce 32; thus, its generation was also disfavored. Thus, the absolute configuration of 33 synthesized via L-proline mediated intramolecular aldol reaction is estimated to be 1R, 4R, 5S, 6R. Although the use of D-proline induced the intramolecular aldol reaction with a conversion yield similar to L-proline, the production ratio of 32 increased (9% NMR yield) and the absolute ee value of ent-33 derived from ent-31 decreased (−85% ee), indicating that the enamine intermediate derived from D-proline may be unsuitable for this reaction.
We established a stereoselective synthesis method for the four contiguous stereogenic centers of the cis-fused 5,6-ring skeleton with the two angular positions in picrotoxane-type sesquiterpenes. This method involves the DL-proline mediated intramolecular aldol reaction of rac-aldehyde 22 with an inner symmetrical 2-methyl-1,3-cyclopentanedione moiety, providing 31 in good yield with high stereoselectivity. We also succeeded in the kinetic resolution of the aldehyde via the use of L-proline, which afforded highly enantiomerically pure 31. Although the intramolecular aldol reaction of ester 10 preferably provided the undesired diastereomer 19, this synthetic methodology could also be developed for the synthesis of other natural products with the similar cis-fused skeleton. Further investigations toward the total synthesis of picrotoxane-type sesquiterpenes including tutin (3) are underway in our laboratory.
All commercially available reagents were used as received. All moisture and air sensitive reactions were carried out in glassware equipped with rubber septa (or a septum) under the positive pressure of argon or nitrogen. When necessary, the glassware was dried under reduced pressure by heating with a heat-gun and solvents were distilled prior to use. The substrates were azeotropically dried if needed by evaporation of their acetonitrile or toluene solution several times to remove trace water that may be contained to the substrates. The reaction mixture was magnetically stirred. Concentration was performed under reduced pressure.
The reactions were monitored by TLC and MS. Anhydrous MgSO4 or Na2SO4 was used to dry organic layers after extraction, and it was removed by filtration through a cotton pad. The filtrate was concentrated and subjected to further purification protocols if necessary. This sequence was represented as “the general drying procedure” in the following experimental methods.
TLC was performed on Merck pre-coated silica gel 60 F-254 plates. Spots were visualized by exposure to UV light, or by immersion into a solution of 2% anisaldehyde, 5% H2SO4 in ethanol or a solution of 10% phosphomolybdic acid in ethanol, followed by heating at approx. 200 °C.
Column chromatography was performed on Kanto Chemical silica gel 60 N (Spherical, neutral, 40–50 or 63–210 µm). The other carrier materials were noted in each case.
The melting points were determined using an AS ONE ATM-02 apparatus and uncorrected. Optical rotations were determined using a JASCO P-2100 polarimeter with a 100 mm cell at 589 nm. IR spectra were recorded on JASCO FT/IR-4100 and Shimadzu IRAffinity-1S with an Attenuated Total Reflection (ATR) sampling unit, and the major absorbance bands are all reported in wavenumbers (cm−1). High-Resolution MS (HRMS) were recorded on a JEOL JMS-T100LC spectrometer at School of Science and Technology, Kwansei Gakuin University and JEOL JMS-T100LP at Global Facility Center, Hokkaido University. The data are reported in units of mass to charge. NMR spectra were recorded on JEOL JNM-ECX-400 (400 MHz for 1H and 101 MHz for 13C) and JNM-ECX-500 (500 MHz for 1H and 126 MHz for 13C) with either TMS or residual proton of deuterated solvent (CDCl3) as internal reference. The 1H-NMR spectroscopic data are indicated by a chemical shift (δ), with the multiplicity, the coupling constants, the integration in parentheses in this order. The multiplicities are abbreviated as s: singlet, d: doublet, t: triplet, q: quartet, m: multiplet, and br: broad. The 13C-NMR spectroscopic data are reported as the chemical shift (δ), with the hydrogen multiplicity obtained from the Distortionless Enhancement by Polarization Transfer (DEPT) spectra in parentheses. The multiplicities are abbreviated as s: C, d: CH, t: CH2, and q: CH3. When the number of the carbon was more than one, the number was added in the parentheses.
Synthesis of 12To a solution of 11 (1.0 g, 8.9 mmol) in H2O (17.8 mL) was added acrolein (750 mg, 13.4 mmol) at room temperature (r.t.). After stirring for 7 d at r.t., to the mixture was added CH2Cl2. The aqueous mixture was extracted with CH2Cl2 and EtOAc. The general drying procedure gave a crude product including 3-(1-methyl-2,5-dioxocyclopentyl)propanal, which was used to the next reaction without further purification.
To a solution of the crude product in toluene (86 mL) was added ethyl 2-(triphenylphosphoranylidene)propionate (3.75 g, 10.3 mmol) at r.t. After stirring for 18 h at r.t., the mixture was concentrated. The residue was purified by silica gel column chromatography (n-hexane/EtOAc = 3/1) to give 12 (1.88 g, 7.45 mmol, 84% yield in 2 steps) as a colorless oil. Data for 12: IR (ATR) ν 2980, 2929, 1716, 1705, 1651, 1454, 1273, 1113, 745 cm−1. 1H-NMR (400 MHz, 25 °C) δ: 6.56 (tq, J = 7.6, 1.4 Hz, 1H), 4.16 (q, J = 7.2 Hz, 2H), 2.87–2.65 (m, 4H), 2.07–2.01 (m, 2H), 1.78–1.74 (m, 2H), 1.76 (d, J = 1.4 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.14 (s, 3H). 13C-NMR (101 MHz, 24 °C) δ: 216.2 (s, 2C), 168.0 (s), 139.7 (d), 129.3 (s), 60.7 (t), 56.5 (s), 35.2 (t, 2C), 33.5 (t), 23.9 (t), 20.0 (q), 14.4 (q), 12.5 (q). HRMS (electrospray ionization (ESI)) m/z [M + Na]+ Calcd for C14H20O4Na 275.1259. Found 275.1257.
Synthesis of 13To a mixture of 12 (1.70 g, 6.73 mmol) and 1,2-bis(trimethylsilyloxy)ethane (7.93 g, 38.4 mmol) in CH2Cl2 (67 mL) was added TMSOTf (300 mg, 1.35 mmol) at −40 °C. The mixture was stirred for 4.5 d at 3 °C. To the mixture was added saturated aqueous sodium hydrogen carbonate. The mixture was extracted with CH2Cl2. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 6/1 to 3/1) to give 13 (1.48 g, 4.35 mmol, 65% yield) as a colorless oil. Data for 13: IR (ATR) ν 2978, 2881, 1703, 1649, 1456, 1368, 1113, 976, 748 cm−1. 1H-NMR (400 MHz, 25 °C) δ: 6.76 (tq, J = 7.6, 1.4 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 4.02–3.94 (m, 4H), 3.92–3.83 (m, 4H), 2.21–2.15 (m, 2H), 1.98–1.86 (m, 4H), 1.82 (d, J = 1.4 Hz, 3H), 1.60–1.53 (m, 2H), 1.28 (t, J = 7.1 Hz, 3H), 1.14 (s, 3H). 13C-NMR (101 MHz, 25 °C) δ: 168.6 (s), 143.6 (d), 127.3 (s), 117.6 (s, 2C), 64.8 (t, 2C), 64.3 (t, 2C), 60.5 (t), 50.2 (s), 32.4 (t, 2C), 28.3 (t), 23.6 (t), 17.1 (q), 14.4 (q), 12.3 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C18H28O6Na 363.1784. Found 363.1781.
Synthesis of 14To a solution of 13 (1.48 g, 4.34 mmol) in THF (43 mL) was added DIBAL (1.03 M in n-hexane, 16.9 mL, 17.4 mmol) at −78 °C. The mixture was stirred for 50 min at −78 °C. After warming to 0 °C, the mixture was additionally stirred for 5 h. To the mixture was added 15% of aqueous Rochelle salt. The mixture was extracted with EtOAc. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 1/2) to give 14 (1.22 g, 4.09 mmol, 95% yield) as a colorless oil. Data for 14: IR (ATR) ν 3763–3076, 2976, 2881, 1462, 1377, 1113, 1057, 974, 949, 727, 667 cm−1. 1H-NMR (400 MHz, 25 °C) δ: 5.41 (dt, J = 7.2, 1.2 Hz, 1H), 4.02–3.93 (m, 6H), 3.91–3.84 (m, 4H), 2.06–2.00 (m, 2H), 1.97–1.89 (m, 4H), 1.66 (d, J = 1.2 Hz, 3H), 1.52–1.47 (m, 2H), 1.30 (br t, J = 5.3 Hz, 1H), 1.13 (s, 3H). 13C-NMR (101 MHz, 25 °C) δ: 134.2 (s), 127.8 (d), 117.7 (s, 2C), 69.3 (t), 64.8 (t, 2C), 64.3 (t, 2C), 50.3 (s), 32.5 (t, 2C), 29.3 (t), 22.4 (t), 17.1 (q), 13.7 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C16H26O5Na 321.1678. Found 321.1673.
Synthesis of 17To a solution of 14 (1.22 g, 4.08 mmol) in MeC(OEt)3 (20 mL) were added PivOH (41.7 mg, 409 µmol) at r.t. After warming to 50 °C, the mixture was stirred for 1 h. After warming to 130 °C, the mixture was additionally stirred for 50 min. After the mixture was cooled to r.t., acetone (20 mL), H2O (20 mL), and 1 M sulfuric acid (5.1 mL) were added. The mixture was stirred for 10.5 h at reflux. After the mixture was cooled to 0 °C, saturated aqueous sodium hydrogen carbonate was added to the mixture. The aqueous mixture was extracted with Et2O. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 10/1 to 5/1) to give 17 (828 mg, 2.95 mmol, 72% yield) as a colorless oil. Data for 17: IR (ATR) ν 3073, 2976, 2939, 1722, 1645, 1454, 1371, 1163, 1031, 897, 669 cm−1. 1H-NMR (400 MHz, 24 °C) δ: 4.73 (dq, J = 1.6, 1.4 Hz, 1H), 4.66 (d, J = 1.6 Hz, 1H), 4.04 (q, J = 7.1 Hz, 2H), 2.78–2.64 (m, 4H), 2.41 (m, 1H), 2.27 (dd, J = 14.5, 7.7 Hz, 1H), 2.21 (dd, J = 14.5, 7.2 Hz, 1H), 1.55 (d, J = 1.4 Hz, 3H), 1.53–1.41 (m, 2H), 1.17 (t, J = 7.1 Hz, 3H), 1.24–1.11 (m, 2H), 1.04 (s, 3H). 13C-NMR (101 MHz, 24 °C) δ: 216.50 (s), 216.45 (s), 172.3 (s), 145.1 (s), 113.2 (t), 60.4 (t), 56.6 (s), 43.9 (d), 39.1 (t), 35.3 (t, 2C), 33.0 (t), 27.2 (t), 19.0 (q), 18.4 (q), 14.3 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C16H24O4Na 303.1572. Found 303.1568.
Synthesis of 10To a solution of 17 (150 mg, 535 µmol) in EtOH (2.0 mL) were added 2-methyl-2-butene (224 mg, 6.42 mmol) and CuBr2 (717 mg, 3.21 mmol) at r.t. The mixture was stirred for 2 h at 50 °C. To the mixture was further added CuBr2 (213 mg, 913 µmol) at 50 °C. After stirring at 50 °C for 20 min, to the mixture were added saturated aqueous sodium hydrogen carbonate and 10% of aqueous sodium thiosulfate. The aqueous mixture was extracted with EtOAc. The combined organic layers were washed with brine. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 9/1 to 8/1) to give 10 (105 mg, 377 µmol, 71% yield) as a yellow oil. Data for 10: IR (ATR) ν 3075, 2978, 2931, 1732, 1701, 1645, 1454, 1371, 1172, 897, 771 cm−1. 1H-NMR (400 MHz, 24 °C) δ: 7.23 (s, 2H), 4.77 (dq, J = 1.8, 1.4 Hz, 1H), 4.70 (d, J = 1.8 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 2.44 (m, 1H), 2.31 (dd, J = 14.8, 8.1 Hz, 1H), 2.25 (dd, J = 14.8, 7.3 Hz, 1H), 1.62–1.49 (m, 2H), 1.57 (d, J = 1.4 Hz, 3H), 1.21 (t, J = 7.1 Hz, 3H), 1.17–1.04 (m, 2H), 1.12 (s, 3H). 13C-NMR (101 MHz, 24 °C) δ: 207.88 (s), 207.85 (s), 172.3 (s), 148.3 (d, 2C), 145.0 (s), 113.1 (t), 60.4 (t), 50.3 (s), 43.9 (d), 39.1 (t), 32.1 (t), 27.5 (t), 19.1 (q), 18.3 (q), 14.3 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C16H22O4Na 301.1416. Found 301.1414.
Synthesis of 18 and 19To a solution of 10 (11.2 mg, 40.2 µmol) in toluene (4.0 mL) were added dropwise KHMDS (0.5 M in toluene, 0.12 mL, 60 µmol) at −78 °C. After stirring for 2 h at −78 °C, to the mixture was added saturated aqueous ammonium chloride at −78 °C. After warming to r.t., the mixture was extracted with EtOAc. The general drying procedure gave a crude product including two diastereomeric aldol products, which was used next reaction without further purification.
To a solution of the crude product in CH2Cl2 (1.0 mL) were added TMSOTf (35.6 mg, 161 µmol) and 2,6-lutidine (41.8 mg, 322 µmol) at 0 °C. After stirring for 30 min at 0 °C, to the mixture was added saturated aqueous ammonium chloride. The mixture was extracted with Et2O. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 20/1) to give a 1 : 1.8 mixture of 18 and 19 (6.8 mg, 19 µmol, 48% yield in 2 steps) as a colorless oil. The diastereomeric mixture was further purified by preparative TLC (n-hexane/EtOAc = 4/1) to give 18 (2.1 mg) as a colorless oil and 19 (4.3 mg) as a colorless oil. Data for 18: IR (ATR) ν 3076, 2937, 1716, 1647, 1456, 1373, 1082, 897, 839, 754 cm−1. 1H-NMR (500 MHz, 22 °C) δ: 8.08 (d, J = 6.3 Hz, 1H), 6.26 (d, J = 6.3 Hz, 1H), 4.70 (d, J = 1.5 Hz, 1H), 4.67 (dq, J = 1.5, 1.2 Hz, 1H), 4.20 (dq, J = 10.5, 7.1 Hz, 1H), 4.07 (dq, J = 10.5, 7.1 Hz, 1H), 3.02 (d, J = 12.8 Hz, 1H), 2.26 (ddd, J = 12.8, 7.3, 7.3 Hz, 1H), 1.70 (d, J = 1.2 Hz, 3H), 1.65–1.60 (m, 2H), 1.48 (m, 1H), 1.38 (m, 1H), 1.26 (t, J = 7.1 Hz, 3H), 1.10 (s, 3H), 0.00 (s, 9H). 13C-NMR (126 MHz, 24 °C) δ: 212.3 (s), 173.3 (s), 161.6 (d), 146.2 (s), 133.6 (d), 112.1 (t), 84.4 (s), 60.5 (t), 53.6 (d), 52.7 (s), 43.4 (d), 33.9 (t), 25.1 (t), 18.9 (q), 17.3 (q), 14.4 (q), 2.36 (q, 3C). HRMS (ESI) m/z [M + Na]+ Calcd for C19H30O4SiNa 373.1811. Found 373.1820. Data for 19: IR (ATR) ν 2980, 2920, 1734, 1647, 1456, 1373, 1174, 899, 852 cm−1. 1H-NMR (500 MHz, 24 °C) δ: 7.75 (d, J = 6.3 Hz, 1H), 6.17 (d, J = 6.3 Hz, 1H), 4.64 (dq, J = 1.7, 1.2 Hz, 1H), 4.54 (d, J = 1.7 Hz, 1H), 4.15 (dq, J = 10.9, 6.9 Hz, 1H), 4.12 (dq, J = 10.9, 6.9 Hz, 1H), 2.63 (ddd, J = 12.0, 8.0, 6.3 Hz, 1H), 2.34 (d, J = 12.0 Hz, 1H), 1.89–1.77 (m, 2H), 1.71 (d, J = 1.2 Hz, 3H), 1.49 (m, 1H), 1.23 (t, J = 6.9 Hz, 3H), 1.14 (m, 1H), 1.05 (s, 3H), 0.09 (s, 9H). 13C-NMR (126 MHz, 23 °C) δ: 209.7 (s), 171.5 (s), 164.3 (d), 148.9 (s), 130.4 (d), 109.2 (t), 83.9 (s), 60.6 (t), 58.6 (d), 54.2 (s), 39.4 (d), 29.2 (t), 26.9 (t), 23.8 (q), 21.9 (q), 14.2 (q), 2.51 (q, 3C). HRMS (ESI) m/z [M + Na]+ Calcd for C19H30O4SiNa 373.1811. Found 373.1815.
Synthesis of 20To a solution of 14 (147 mg, 493 µmol) in toluene (2.5 mL) was added N,N-dimethylacetamide dimethyl acetal (stabilized with 5–10% MeOH, 228 mg, 205–217 mg as N,N-dimethylacetamide dimethyl acetal, 1.81–1.92 mmol) at r.t. The mixture was stirred at reflux for 23 h. After concentration of the mixture, the resulting residue was purified by silica gel column chromatography (n-hexane/EtOAc = 2/1 to 1/3) to give 20 (137 mg, 372 µmol, 76% yield) as a yellow oil. Data for 20: IR (ATR) ν 2978, 2945, 2881, 1640, 1395, 1065, 749 cm−1. 1H-NMR (500 MHz, 20 °C) δ: 4.73 (s, 1H), 4.70 (s, 1H), 4.00–3.91 (m, 4H), 3.87–3.82 (m, 4H), 2.99 (s, 3H), 2.91 (s, 3H), 2.51 (br m, 1H), 2.42–2.30 (m, 2H), 1.94–1.82 (m, 4H), 1.67 (s, 3H), 1.43–1.34 (m, 4H), 1.07 (s, 3H). 13C-NMR (126 MHz, 21 °C) δ: 172.3 (s), 147.4 (s), 117.74 (s), 117.71 (s), 111.3 (t), 64.9 (t), 64.7 (t), 64.3 (t), 64.2 (t), 50.2 (s), 44.5 (d), 38.3 (t), 37.6 (q), 35.5 (q), 32.6 (t), 32.4 (t), 27.2 (t), 27.0 (t), 19.4 (q), 17.2 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C20H33O5NNa 390.2251. Found 390.2245.
Synthesis of 21To a solution of 20 (447 mg, 1.22 mmol) in THF (4.0 mL) were added Cp2ZrCl2 (427 mg, 1.46 mmol) and LiAlH(Ot-Bu)3 (1.0 M in THF, 1.3 mL, 1.3 mmol) at 0 °C. After stirring for 1 h at 0 °C, to the mixture were added saturated aqueous Rochelle salt and Et2O. The mixture was extracted with Et2O. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 7/1 to 3/1) to give 21 (271 mg, 836 µmol, 68% yield) as a pale yellow oil. Data for 21: IR (ATR) ν 2977, 2948, 2881, 1722, 1069, 772 cm−1. 1H-NMR (500 MHz, 20 °C) δ: 9.65 (br t, J = 1.7 Hz, 1H), 4.77 (s, 1H), 4.74 (s, 1H), 3.99–3.93 (m, 4H), 3.89–3.81 (m, 4H), 2.56 (br m, 1H), 2.42 (dt, J = 7.5, 1.7 Hz, 2H), 1.94–1.82 (m, 4H), 1.64 (s, 3H), 1.42–1.36 (m, 4H), 1.07 (s, 3H). 13C-NMR (126 MHz, 21 °C) δ: 202.9 (d), 146.2 (s), 117.6 (s, 2C), 112.4 (t), 64.8 (t), 64.7 (t), 64.2 (t, 2C), 50.1 (s), 47.6 (t), 42.7 (d), 32.5 (t), 32.4 (t), 27.3 (t), 27.0 (t), 18.9 (q), 17.1 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C18H28O5Na 347.1829. Found 347.1822.
Synthesis of 22To a stirred solution of 21 (36.5 mg, 113 µmol) in acetone (1.0 mL) was added 1 M sulfuric acid (100 µL) at r.t. After stirring at 40 °C for 17 h, to the mixture was added saturated aqueous sodium hydrogen carbonate at r.t. The mixture was extracted with EtOAc. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 5/1 to 3/1) to give 22 (15.0 mg, 63.5 µmol, 56% yield) as a colorless oil.
Data for 22: IR (ATR) ν 2967, 2936, 1716, 1452, 1056, 897 cm−1. 1H-NMR (500 MHz, 19 °C) δ: 9.62 (dd, J = 2.3, 1.7 Hz, 1H), 4.82 (s, 1H), 4.75 (s, 1H), 2.83–2.68 (m, 4H), 2.56 (m, 1H), 2.42 (ddd, J = 16.4, 8.5, 2.3 Hz, 1H), 2.34 (ddd, J = 16.4, 6.3, 1.7 Hz, 1H), 1.62–1.50 (m, 2H), 1.59 (s, 3H), 1.23–1.17 (m, 2H), 1.10 (s, 3H). 13C-NMR (126 MHz, 20 °C) δ: 216.54 (s), 216.47 (s), 201.8 (d), 144.8 (s), 113.6 (t), 56.6 (s), 47.4 (t), 41.6 (d), 35.3 (t, 2C), 32.7 (t), 27.5 (t), 19.4 (q), 18.5 (q). HRMS atmospheric pressure chemical ionization m/z [M − H]− Calcd for C14H19O3 235.1340. Found 235.1337.
Synthesis of 31 via the Use of DL-ProlineTo a solution of 22 (18.8 mg, 79.6 µmol) in THF (1.5 mL) and DMSO (150 µL) was added DL-proline (45.8 mg, 398 µmol) at 0 °C. After stirring for 48 h at 0 °C, to the mixture was added brine. The mixture was extracted with EtOAc. The combined organic layer was successively washed with brine. The general drying procedure afforded a crude product including a 49 : 1 mixture of 31 and 32. The crude solid product was purified by trituration with toluene/n-hexane (1/1) to give 31 (13.7 mg, 58.0 µmol, 73% yield) as a white solid. Data for 31: mp 121–124 °C. IR (ATR) ν 3438, 2973, 2936, 1719, 1376, 1071, 952 cm−1. 1H-NMR (500 MHz, 20 °C) δ: 9.77 (s, 1H), 4.94 (br s, 1H), 4.86 (br s, 1H), 2.84 (br s, 1H), 2.57–2.52 (m, 2H), 2.51–2.39 (m, 2H), 2.30 (m, 1H), 1.95 (m, 1H), 1.79 (s, 3H), 1.73 (m, 1H), 1.55 (m, 1H), 1.45–1.40 (m, 2H), 1.11 (s, 3H). 13C-NMR (126 MHz, 20 °C) δ: 219.1 (s), 207.1 (d), 145.3 (s), 113.6 (t), 80.8 (s), 55.3 (d), 53.4 (s), 44.3 (d), 32.9 (t), 31.5 (t), 28.5 (t), 26.6 (t), 20.2 (q), 12.9 (q). HRMS (ESI) m/z [M − H]− Calcd for C14H19O3 235.1340. Found 235.1337.
Synthesis of 32 via the Use of AluminaTo a solution of 22 (27.3 mg, 116 µmol) in toluene (1.0 mL) was added alumina (200 mg) at r.t. After stirring for 1.5 h at r.t., the mixture was filtered and concentrated to give a crude product including a 21 : 1 mixture of 32 and 31 (50% NMR yield). The residue was purified by silica gel column chromatography (n-hexane/EtOAc = 8/1 to 4/1) to give 32 (8.4 mg, 36 µmol, 31% yield) as a colorless oil and a mixture of 32 and side products. Data for 32: 1H-NMR (500 MHz, 16 °C) δ: 9.64 (d, J = 2.9 Hz, 1H), 4.79 (br s, 1H), 4.78 (br s, 1H), 2.86 (br s, 1H), 2.67 (ddd, J = 12.0, 12.0, 2.9 Hz, 1H), 2.57 (dd, J = 20.1, 10.3 Hz, 1H), 2.34–2.19 (m, 2H), 2.06–1.97 (m, 3H), 1.65 (s, 3H), 1.59 (m, 1H), 1.45 (m, 1H), 1.17 (m, 1H), 1.00 (s, 3H). 13C-NMR (126 MHz, 17 °C) δ: 217.5 (s), 206.9 (d), 145.3 (s), 113.2 (t), 79.3 (s), 56.1 (d), 53.9 (s), 42.4 (d), 34.5 (t), 30.9 (t), 28.7 (t), 28.5 (t), 19.6 (q), 19.2 (q).
Synthesis of rac-33 and 34To a solution a 1.9 : 1 mixture of 31 and 32 (5.5 mg, 23 µmol) in THF (300 µL), t-BuOH (300 µL), H2O (300 µL), and 2-methyl-2-butene (100 µL) were added NaClO2 (6.8 mg, 75 µmol) and NaH2PO4·2H2O (12.0 mg, 76.9 µmol) at 0 °C. After stirring for 2.5 h at 0 °C, to the mixture was added NaSO3·7H2O then 1 M hydrochloric acid. The mixture was extracted with EtOAc. The general drying procedure afforded a crude product including two carboxylic acids, which was used to the next reaction without further purification.
To a solution of the crude product in DMF (500 µL) were added K2CO3 (6.0 mg, 47 µmol) and BnBr (6.0 mg, 35 µmol) at r.t. After stirring for 14 h at r.t., to the mixture was added saturated aqueous ammonium chloride. The mixture was extracted with EtOAc. The combined organic layer was successively washed with brine. After the general drying procedure, the residue was purified by preparative TLC (n-hexane/EtOAc = 4/1) to give 33 (3.1 mg, 9.1 µmol, 39% yield in 2 steps) as a white solid and 34 (1.7 mg, 5.0 µmol, 21% yield in 2 steps) as a white solid. Data for 33: mp 97–100 °C. IR (ATR) ν 3473, 3028, 2967, 2934, 1741, 1701, 1164, 901 cm−1. 1H-NMR (500 MHz, 19 °C) δ: 7.39–7.33 (m, 5H), 5.15 (d, J = 12.0 Hz, 1H), 5.12 (d, J = 12.0 Hz, 1H), 4.75 (br s, 2H), 2.79 (d, J = 12.6 Hz, 1H), 2.51–2.33 (m, 4H), 2.13 (br s, 1H), 1.74–1.68 (m, 5H), 1.42–1.37 (m, 3H), 1.10 (s, 3H). 13C-NMR (126 MHz, 18 °C) δ: 219.7 (s), 172.4 (s), 146.6 (s), 135.6 (s), 128.8 (d, 2C), 128.71 (d, 2C), 128.65 (d), 111.5 (t), 80.6 (s), 67.0 (t), 53.6 (s), 53.0 (d), 45.1 (d), 33.2 (t), 31.8 (t), 29.0 (t), 26.3 (t), 20.8 (q), 13.0 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C21H26O4Na 365.1723. Found 365.1721. HPLC (DAICEL CHIRALPAK AD-H, n-hexane/i-PrOH = 90/10, flow rate = 1.0 mL/min, 254 nm): tR = 12.8, 16.3 min. Data for 34: mp 120–124 °C. IR (ATR) ν 3501, 3032, 2940, 1732, 1165, 772 cm−1. 1H-NMR (500 MHz, 20 °C) δ: 7.39–7.30 (m, 5H), 5.14 (d, J = 12.0 Hz, 1H), 5.06 (d, J = 12.0 Hz, 1H), 4.68 (br s, 1H), 4.59 (br s, 1H), 4.57 (br s, 1H), 2.62–2.52 (m, 2H), 2.27 (d, J = 12.0 Hz, 1H), 2.26 (m, 1H), 2.13 (m, 1H), 2.00 (m, 1H), 1.78 (dd, J = 12.6, 9.7 Hz, 1H), 1.55 (s, 3H), 1.55–1.44 (m, 2H), 1.17 (m, 1H), 1.04 (s, 3H). 13C-NMR (126 MHz, 20 °C) δ: 217.7 (s), 175.0 (s), 146.1 (s), 135.1 (s), 128.9 (d, 2C), 128.76 (d), 128.7 (d, 2C), 112.8 (t), 77.2 (s), 67.2 (t), 53.3 (s), 52.3 (d), 43.9 (d), 34.6 (t), 31.5 (t), 28.3 (t), 28.1 (t), 19.6 (q), 18.8 (q). HRMS (ESI) m/z [M + Na]+ Calcd for C21H26O4Na 365.1723. Found 365.1719.
Synthesis of Optically Active 33 via the Use of L-ProlineTo a solution 22 (41.5 mg, 176 µmol) in THF (3.4 mL) and DMSO (340 µL) was added L-proline (101 mg, 878 µmol) at 0 °C. After stirring for 44 h at 0 °C, to the mixture was added brine. The mixture was extracted with EtOAc. The combined organic layer was successively washed with brine. The general drying procedure afforded a crude product including a 12 : 1 mixture of 31 and 32 (41% NMR yield) and 22 (20% NMR yield). The crude product was used to the next reaction without further purification.
To a solution the crude product in THF (800 µL), t-BuOH (800 µL), H2O (800 µL), and 2-methyl-2-butene (400 µL) were added NaClO2 (54.5 mg, 603 µmol) and NaH2PO4·2H2O (90.1 mg, 578 µmol) at 0 °C. After stirring for 2 h at 0 °C, to the mixture was added NaSO3·7H2O then 1 M hydrochloric acid. The mixture was extracted with EtOAc. The general drying procedure afforded a crude product, which was used to the next reaction without further purification.
To a solution of the crude product in DMF (2.0 mL) were added K2CO3 (49.7 mg, 360 µmol) and BnBr (44.7 mg, 264 µmol) at r.t. After stirring for 30 min at r.t., to the mixture was added saturated aqueous ammonium chloride. The mixture was extracted with Et2O. The combined organic layer was washed with brine. After the general drying procedure, the residue was purified by silica gel column chromatography (toluene/CH2Cl2 = 1/1 to 0/1, then CH2Cl2/EtOAc = 15/1 to 8/1) to give an impure product 33. The product was further purified by trituration with n-hexane to give 33 (8.3 mg, 24 µmol, >99% ee, 14% yield in 3 steps) as a white solid. Additional data: [α]D27 +14.7 (c 0.280, CHCl3). HPLC (DAICEL CHIRALPAK AD-H, n-hexane/i-PrOH = 90/10, flow rate = 1.0 mL/min, 254 nm): tmajor = 13.0 min.
Synthesis of ent-33 via the Use of D-ProlineTo a solution 22 (26.8 mg, 113 µmol) in THF (2.2 mL) and DMSO (220 µL) was added D-proline (67.5 mg, 586 µmol) at 0 °C. After stirring for 40 h at 0 °C, to the mixture was added brine. The mixture was extracted with EtOAc. The combined organic layer was successively washed with brine. The general drying procedure afforded a crude product including a 3.3 : 1 mixture of ent-31 and ent-32 (40% NMR yield) and 22 (13% NMR yield). The crude product was used to the next reaction without further purification.
To a solution the crude product in THF (500 µL), t-BuOH (500 µL), H2O (500 µL), and 2-methyl-2-butene (200 µL) were added NaClO2 (41.7 mg, 461 µmol) and NaH2PO4·2H2O (60.3 mg, 387 µmol) at 0 °C. After stirring for 2.5 h at 0 °C, to the mixture was added NaSO3·7H2O then 1 M hydrochloric acid. The mixture was extracted with EtOAc. The general drying procedure afforded a crude product, which was used to the next reaction without further purification.
To a solution of the crude product in DMF (1.5 mL) were added K2CO3 (31.8 mg, 248 µmol) and BnBr (28.8 mg, 170 µmol) at r.t. After stirring for 1.5 h at r.t., to the mixture was added saturated aqueous ammonium chloride. The mixture was extracted with Et2O. The combined organic layer was washed with brine. After the general drying procedure, the residue was purified by silica gel column chromatography (n-hexane/EtOAc = 15/1 to 4/1) to give an impure product ent-33. The product was further purified by preparative TLC (CH2Cl2/EtOAc = 15/1) to give ent-33 (6.3 mg, 18.4 µmol, −85% ee, 16% yield in 3 steps) as a white solid. Additional data: [α]D26 −8.42 (c 0.260, CHCl3). HPLC (DAICEL CHIRALPAK AD-H, n-hexane/i-PrOH = 90/10, flow rate = 1.0 mL/min, 254 nm): tmajor = 16.5 min; tminor = 12.9 min.
This research was funded by JSPS KAKENHI, Grant numbers JP19K15549, JP21H01923, JP21K14616, and JP16H01163 in Middle Molecular Strategy.
The authors declare no conflict of interest.
This paper is dedicated to the memory of Prof. Dr. Toshiyuki Kan who sadly passed away on July 23, 2021.
This article contains supplementary materials.