Introduction
Isoxazolidine contains an N–O bond in a five-membered cycle and is an important heterocycle for the design of biologically active molecules owing to the high fraction of sp3 carbon atoms.1) As an efficient reaction to form two bonds in one step, catalytic asymmetric (3 + 2) cycloaddition (1,3-dipolar cycloaddition) is commonly used to prepare this architecture.2) Our interest in this molecule and this reaction is to explore an asymmetric catalytic strategy that yields structurally unique isoxazolidines. Specifically, we have explored the inverse-electron-demand (3 + 2) cycloaddition (i.e., a type-III cycloaddition according to Sustmann’s classification)3,4) using α-keto ester enolates as formal 1,3-dipolarophiles5–7) on the basis of the ambiphilic nature of α-keto esters 1, which inherently possess nucleophilic and electrophilic reactivity.8,9) The reaction is initiated by the Mannich-like addition of an α-keto ester enolate to a nitrone,10) which results in the formation of two adjacent stereocenters. Then, the diastereoselective cyclization spontaneously occurs to form tert-butyl 5-hydroxyisoxazolidine-5-carboxylate (5-hydroxyisoxazolidine) containing a hemiketal with three contiguous stereocenters. Stereochemically well-defined 5-hydroxyisoxazolidines have been used for several types of downstream transformations to yield a range of chiral building blocks, including cyclic and open-chained forms.7,11–13)
During our studies, we encountered a unique diastereodivergence14–17) in the (3 + 2) cycloaddition using a Ni(II) complex prepared from Ni(OAc)2·4H2O and (R,R)-diamine 35,7) (Chart 1). Briefly, the reaction with cyclic (E)-nitrones 2 afforded anti,anti-adducts 4.5) In this reaction, the use of 3a having cyclohexyl groups in 3 is essential to attain high stereoselectivity. When we employed the reaction with typical π-conjugated (Z)-nitrones,5,7) the corresponding cycloaddition adducts were not detected, probably because of the unfavorable steric repulsion between the (Z)-nitrone and diamine ligand. In marked contrast, the reaction with the E/Z-isomerizable C-CN nitrones 5 resulted in the formation of syn,anti-6 as the major adduct, although each geometrical isomer of 5 has a similar gap between the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO) to those of the other nitrones that we investigated. This result indicates that the nitrile group in 5 plays a crucial role in selectively enhancing the syn-addition.7) Further, the catalytic system works well even with simple substrates and a diamine ligand. For example, the reaction between 1a (R1 = Me, R2 = tBu) and 5a (R3 = Me) using the simple ligand (R,R)-3c (R = Me) under standard conditions [Ni(OAc)2·4H2O/(R,R)-3c (10 mol%), 5a (1.2 equivalent), 0 °C] afforded 6aa (R1 = Me, R2 = tBu, and R3 = Me) with comparably high diastereo- and enantioselectivities.7)
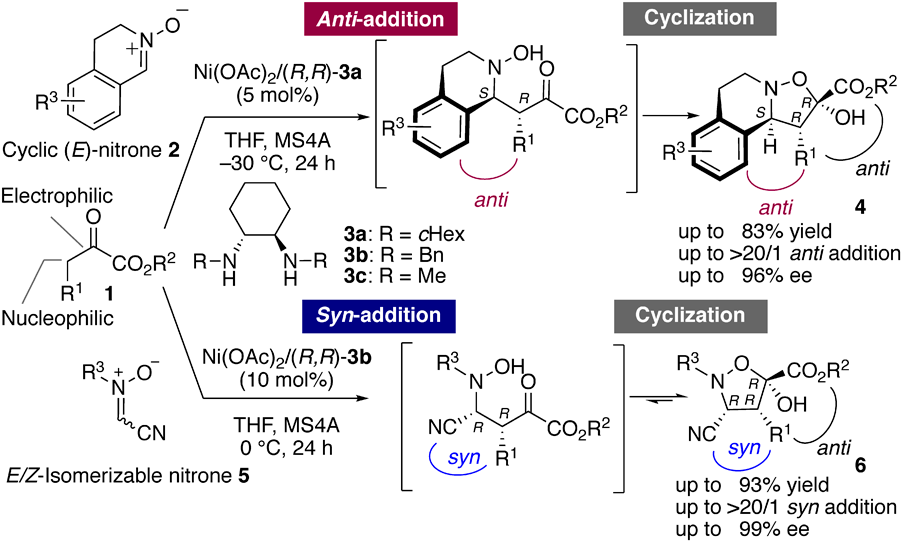
Chart 1. Substrate-Dependent Diastereodivergence in Catalytic Asymmetric (3 + 2) Cycloaddition of 1 Using a Ni(II)–Diamine–Acetate Catalyst
In addition to these experiments, we used density functional theory (DFT) calculations to identify the key catalytic parameter determining the rate- and stereo-determining step(s) (Fig. 1). Specifically, our recent studies on the (3 + 2) cycloaddition of 1a (R1 = Me, R2 = tBu) and E/Z-isomerizable 5a (R3 = Me) with (R,R)-3c as the ligand (Fig. 1-i)5) have revealed that the induced-fit-like deformation of 5a upon binding to the Ni(II)–enolate is key in the syn-addition process (Figs. 1-ii and 1-iii). In the lowest-energy transition state (TS, TS-II-A-Z/Z), the (Z)-form of 5a is stabilized by non-classical attractive interactions (C–H/N and C–H/π interactions), leading to the selective enhancement of the syn-addition reaction in accordance with the empirical results (Fig. 1-iii). However, to date, computational surveys have only focused on the TSs for the reaction of the C-CN nitrone 5a, and the TSs in the reaction with cyclic (E)-nitrone 2 have not yet been analyzed, although we have proposed a TS model that allows the synergistic activation of both the enolate and nitrone in asymmetric space on the basis of electron-density distribution (EDD) analysis18,19) of the Ni(II) complex (CCDC 1482741) in the ground state. Herein, we present the results of our computational investigation of the (3 + 2) cycloaddition of the Ni(II)–enolate with cyclic (E)-nitrone as a model for comparison with the E/Z-isomerizable C-CN nitrone. Specifically, we describe our evaluation of the energy level diagrams, TS characterization, molecular orbital analysis, and distortion and interaction analysis. These computational findings explain how different stereoselective outcomes can be attained by simply alternating the structure of the nitrone in an acid/base mechanism via a common chemical space.
Results and Discussion
Computational chemistry is a powerful tool to gain insights into TSs on a reasonable time scale, even for transition metal catalysts. However, identifying the lowest-energy TS among the possible reaction pathways in transition metal catalysis remains a significant challenge, even with empirical mechanistic evidence. Because most catalytically active species are undetectable as a result of their short lifetimes, the energy differences between multiple potential pathways must be analyzed carefully. In addition, finding a suitable functional/basis set also requires considerable effort, especially when dispersion interactions must be considered.20–23) This situation is indeed the case in the field of asymmetric nickel catalysis.24–29) Although the number of synthetic reactions using chiral Ni complexes has been growing steadily in recent years,24–28) DFT calculations of asymmetric acid–base catalysis with Ni complexes have seldom been carried out.29) To characterize the TSs in open-shell Ni-catalyzed systems, we have employed a combined experimental and computational approach,5–7) in which the simulated spectra of the catalyst, reactants, and products in the ground state are assessed by comparing various experimentally obtained spectra.30,31) For example, on the basis of a series of earlier structural analyses [IR, UV, and electronic circular dichroism (ECD)] in tetrahydrofuran (THF), we concluded that Ni(II)–3a–(OAc)2 is more stable in the triplet state than the singlet state.5) Crucially, DFT calculations of Ni(II)–3a–(OAc)2 in the triplet configuration reproduced the features in the spectroscopic analyses, including (i) the unique carboxylate shifts (1598 and 1558 cm−1) in the IR spectrum, (ii) broad bands of pseudo-octahedral structure of Ni(d8) (408, 753, and 1182 nm) in the UV/Vis spectrum, and (iii) strong intensities in the near-IR region of the ECD spectra.5)
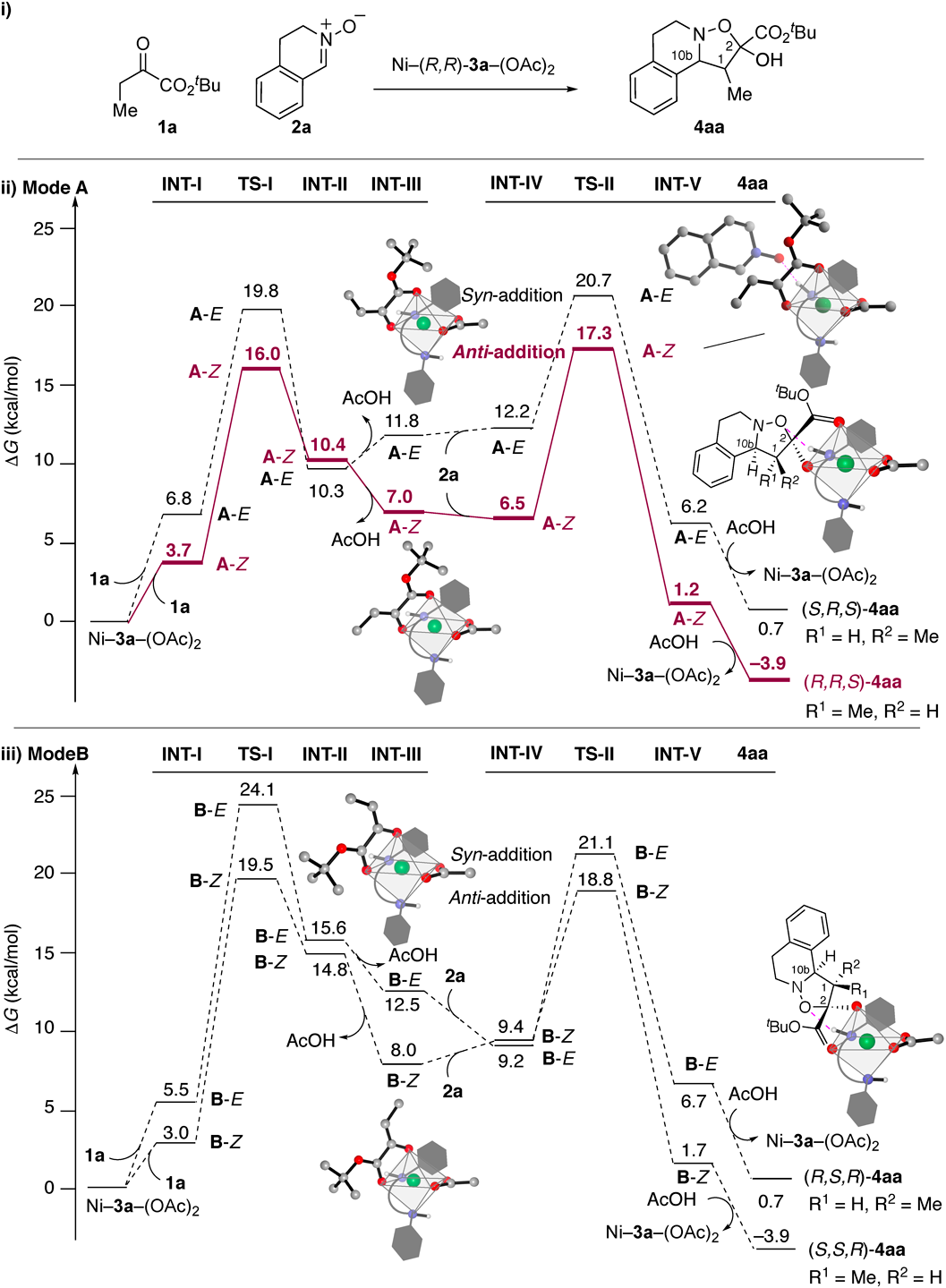
Building on our previous reports,5,7) we began this computational study (Fig. 2) by evaluating the energy diagrams of the (3 + 2) cycloaddition of 1a (R1 = Me and R2 = tBu) and 2a (R3 = H) with (R,R)-3a (R = cHex) (Fig. 2-i) at the UM06/6-311g(d,p)/CPCM(THF) (Ni: SDD)//UωΒ97XD/def2-SVPP (Ni: LanL2DZ) level of theory using Gaussian 16.32–35) Considering that dissociative ligand exchange is possible in this catalytic system, our computational analysis contrasts the two main reaction coordinates, for which the ketone carbonyl in the ester is placed in different positions.5,7) The ketone carbonyl in the α-keto ester coordinates to the metal center at the equatorial position in Mode A (Fig. 2-ii), whereas the ketone carbonyl is placed at the pseudoapical position in Mode B (Fig. 2-iii). Another key force in our proposed TS model5,7) is the outer-sphere hydrogen bonding with the diamine ligand, which enables to control the reaction face of nitrones.36–39) The carbon–carbon bond-formation should be taken place from the Si-face in 2a in Mode A, while the reaction should occur from the Re-face in Mode B. Each energy barrier for the reaction from the opposite face in 2a is considerably large. Thus, additional consideration of the possible geometrical isomerization of the enolates in Mode A and Mode B led us to examine four reaction pathways. In this mechanistic scenario, the coordination mode of the α-keto ester enolate regulates the stereochemistry at the C(10b) and C(2) positions in 4aa, whereas those at the C(1) position in 4aa should be controlled by the geometry of the double bond in the enolate.
As we reported previously, Ni(II)–(R,R)-3a–(OAc)2 (CCDC 1482741) is uniquely distorted. One acetate that coordinates with the metal center in the apical-equatorial coordination mode is more labile than the other acetate that binds to the metal center in the equatorial-equatorial mode.5) Thus, we computed the enolate formation process, wherein the labile acetate anion acts as Brønsted base to enhance the deprotonation of α-proton in 1a with Lewis acidic activation of the ketone in 1a, generating Ni(II)–enolate (Supplementary Figs. S1–S4). The trend in the energies of enolate formation (INT-I, TS-I, INT-II, and INT-III) with (R,R)-3a (R = cHex) is broadly similar to that reported previously for (R,R)-3c (R = Me).7) As discussed, the 1,3-allylic strain40) should increase the energies of the TSs (TS-I-A-E and TS-I-B-E) in the formation of the (E)-Ni(II)–enolates (INT-III-A-E and INT-III-B-E). The pathway to form the (Z)-Ni–enolate in Mode A is predicted to be both kinetically (TS-I-A-Z: 16.0 kcal/mol and TS-I-B-Z: 19.5 kcal/mol) and thermodynamically preferred (INT-III-A-Z: 7.0 kcal/mol and INT-III-B-Z: 8.0 kcal/mol). The kinetic bias in TS-I-B-Z compared to TS-I-A-Z may arise from the unfavorable steric repulsion between reactants and ligand (Supplementary Fig. S2). The geometrical distortion induced in Mode A, in which the bonds lying along the z-axis are elongated compared to other axes, also helps explain thermodynamic stability of INT-III-A-Z compared to INT-III-B-Z (Supplementary Fig. S4), as seen in the structural features of Ni(II)–(R,R)-3a–(OAc)2 (CCDC 1482741). We also predicted that even the most favorable intermediate, INT-III-A-Z (7.0 kcal/mol), would be more energetic than the reference point [Ni(II)–(R,R)-3a–OAc)2]. These results suggest that isolating the Ni(II)–enolate–diamine complex would be difficult even if sterically demanding cyclohexyl groups are incorporated into the side arm in 3.
The three contiguous stereocenters in 4aa are formed in TS-II without generating the Mannich intermediates in the formal (3 + 2) cycloaddition (INT-IV, TS-II, and INT-V). Owing to the constrained nature of cyclic (E)-nitrone 2a, the nitrogen inversion41) in INT-Vs was not observed, unlike the reaction of α-keto ester 1a with C-CN nitrone 5a.7) The energetic sequence is similar to that in the reaction with E/Z-isomerizable 5a. A critical difference between the cyclic (E)-nitrone 2a and the E/Z-isomerizable C-CN nitrone 5a lies in the energy balance between the TS-Is and TS-IIs. The lowest TS of the formal (3 + 2) cycloaddition (TS-II-A-Z: 17.3 kcal/mol) is calculated to be higher in energy than that of the deprotonation (TS-I-A-Z: 16.0 kcal/mol). These results suggest that TS-II is the rate-, enantio-, and diastereo (addition/cyclization)-determining step in the reaction with 2a. Importantly, as we previously proposed based on the EDD analysis,5) TS-II-A-Z was indeed identified as the lowest-energy TS for the formal (3 + 2) cycloaddition with 2a. The second lowest TS, TS-II-B-Z (18.8 kcal/mol), which leads to the minor enantiomer [(1S,2S,10bR)-4aa], was predicted to be disfavored compared to TS-II-A-Z (17.3 kcal/mol) by 1.5 kcal/mol. In the lowest-energy pathway, which is initiated by the generation of a (Z)-Ni(II)–enolate in Mode A, (1R,2R,10bS)-4aa (ΔG = –3.9 kcal/mol) is released with the regeneration of Ni(II)–(R,R)-3a–(OAc)2. This pathway turned out to be both kinetically and thermodynamically favorable, consistent with the experimental observations.5)
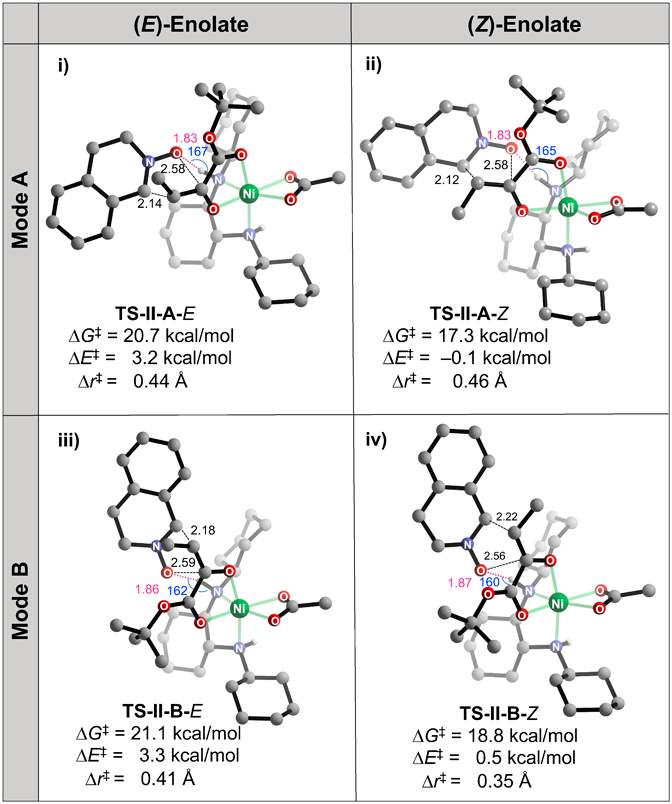
The outer-sphere hydrogen-bond activation of cyclic (E)-nitrone 2a36–39) should play a crucial role in fixing the two reactants in the rate- and stereo-determining TS-IIs (Fig. 3). The directional hydrogen-bonding interactions between the N-H group in the ligand and 2a are conserved in each TS-II [d(H···O): 1.83–1.87 and ∠NHO = 160–167°]. Characteristically, the length of the C–C bond is shorter than that of the C–O bonds; the asynchronous bond-forming process can be explained by assuming that the Ni(II)–enolate acts as a nucleophile.5–7) Specifically, the value of the asynchronicity (Δr‡ = Δr‡C−O−Δr‡C−C) in the lowest TS (TS-II-A-Z: 0.46 Å) is the highest of the four TSs (TS-II-A-E: 0.44 Å, TS-II-B-E: 0.41 Å, and TS-II-B-Z: 0.35 Å).
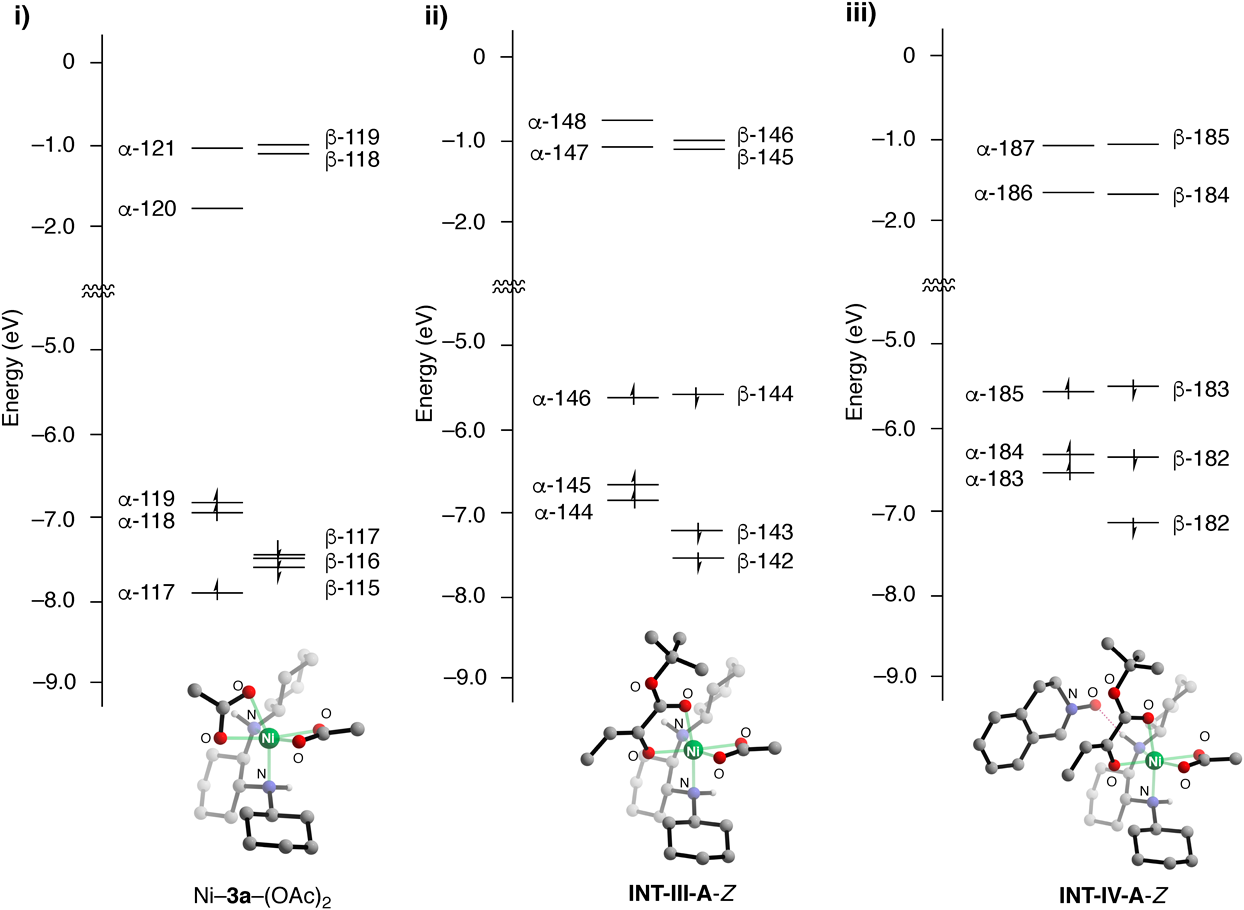
Next, we characterized the molecular orbitals in INT-III-A-Z (Ni–enolate) (Figs. 4, 5). As mentioned, the splitting of the d-orbitals in Ni(II)–3a–(OAc)2, which contains triplet singly occupied molecular orbitals (SOMOs), observed in the experimental spectra was reproduced well5) (Fig. 4-i). In contrast, two electrons having similar energies (α-146: −5.6 eV and β-144: −5.5 eV) are predicted to be energetically higher lying than the SOMO (α-145: −6.6 eV), suggesting that these two electrons act as a nucleophile, even in the triplet configuration7) (Fig. 4-ii). The situation is different from the conventional electronic configuration in the transition metal complex because the SOMO typically has higher energy than the HOMO according to the Aufbau principle (building-up principle). Several examples of SOMO–HOMO level inversions (SHI), in which the energy of the SOMO is lower than that of the doubly occupied HOMO, have been documented.42) However, of note, the occurrence of SHI in Ni(II) complexes has only recently been reported.43) Jeong et al. characterized the SHI property in high-spin Ni(II) complexes, which were prepared from Ni(NO3), 1,4,7-trimethyl-1,4,7-triazacyclononane, and 3-hydroxyflavone, as a functional model of quercetin 2,4-dioxygenases. On the basis of spectroscopic analyses, together with DFT calculation, they revealed that the electron of HOMO in their high-spin Ni(II) complex is delocalized in the flavonolate ligand in the triplet configuration. The SHI property in the Ni(II) complex suggests that flavonolate is preferentially reacted with air to promote oxygenation rather than the oxidation of the Ni(II).43) In our case, the energies of the HOMO-like electrons in the α-SOMO (α-185: −5.5 eV) and β-SOMO (β-183: −5.4 eV) are stabilized only slightly upon binding the cyclic (E)-nitrone 2a in INT-IV-A-Z (Figs. 4-iii, 5-ii) relative to the corresponding HOMO-like electrons in INT-III-A-Z (Fig. 4-ii). These results not only shed light on a new aspect of the Ni(II) complex with the SHI configuration as a chiral nucleophile to promote the catalytic asymmetric C–C bond-formation but also highlight that Sustmann’s classification can be extended to Ni(II)-catalyzed (3 + 2) cycloaddition systems in the triplet configuration.3,4)
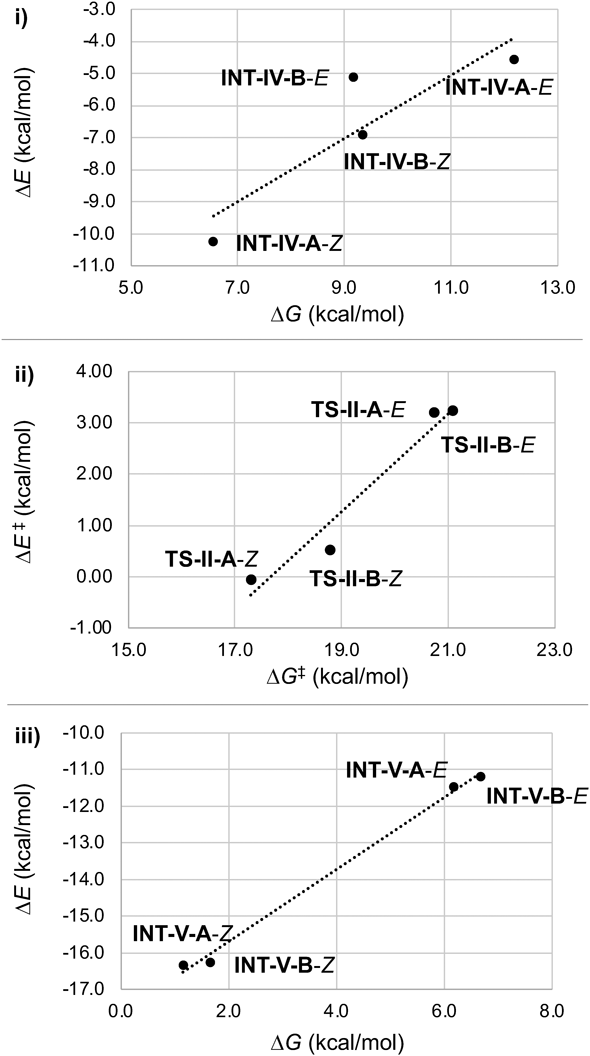
The relationship between ΔG‡ and ΔE‡ in the formal (3 + 2) cycloaddition reaction of cyclic (E)-nitrone 2a is broadly similar to that of 5a.7) Each value of ΔG‡ (TS-II-A-E: 20.7 kcal/mol, TS-II-A-Z: 17.3 kcal/mol, TS-II-B-E: 21.1 kcal/mol, and TS-II-B-Z: 18.8 kcal/mol) is significantly larger than the corresponding ΔE‡ (TS-II-A-E: 3.2 kcal/mol, TS-II-A-Z: −0.1 kcal/mol, TS-II-B-E: 3.3 kcal/mol, and TS-II-B-Z: 0.5 kcal/mol). Further, on plotting ΔG and ΔE for INT-IV, TS-II, and INT-V, linear relationships were obtained (Fig. 6). These results suggest that the energy balance between ΔG and ΔE during each bond-forming process is well correlated. The formation of two bonds in the Ni(II)-catalyzed (3 + 2) cycloaddition reflects the large entropic penalty in ΔG, whereas the ternary and binary complexes (INT-IVs, TS-IIs, and INT-Vs) are electronically stable.
Having characterized the key structures including Ni–3a–(OAc)2, INT-III, and INT-V, and TSs, we finally conducted a distortion–interaction (or activation strain) analysis of the TSs to gain insight into the structural changes during the bond-forming process44–48) (Table 1). In this analysis, the interaction energies (ΔE‡int) are obtained by subtracting the computationally calculated activation energies (ΔE‡act) from the total ΔE‡dist [=ΔE‡dist (Ni–enolate) + ΔE‡dist (nitrone)]. In our previous work (entry 1), the negative activation energy (ΔE‡act=−1.8 kcal/mol) and the deformation of 5a [ΔE‡dist (nitrone) = 10.2 kcal/mol], which is assisted by C–H/N and C–H/π interactions between 5a and ligand 3c (Fig. 1-iii), cooperatively contribute to the large interaction energy (ΔE‡int = −17.1).7) On the other hand, in the lowest-energy TS (TS-II-A-Z) for the reaction of 2a, the value [ΔE‡dist (nitrone) = 8.8 kcal/mol] is significantly smaller. Moreover, the interaction energy [ΔE‡int = −15.0 kcal/mol] in the lowest-energy TS (TS-II-A-Z; entry 3) is also smaller than that of the second lowest-energy TS, TS-II-B-Z [entry 5; ΔE‡int = −16.0 kcal/mol] in the reaction of cyclic (E)-nitrone 2a. Thus, the structural distortion of 2a is not the primary driving force for anti-addition. The minimal energy values in INT-IV-A-Z (ΔG = 6.5 kcal/mol) and INT-V-A-Z (ΔG = 1.2 kcal/mol), which are shown in Fig. 2-ii, also suggest that the lowest-energy transition structure TS-II-A-Z should be more geometrically similar to the reactant and product of all TS-IIs.
Table 1. Distortion and Interaction Analysis
a)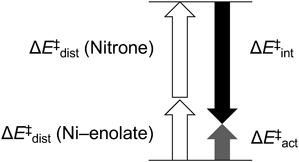 |
|---|
| Entry | Nitrone | TS-II | ΔE‡dist (Ni–enolate) | ΔE‡dist (nitrone) | ΔE‡dist | ΔE‡act | ΔE‡int | Δr‡b) |
|---|
| 1c) | 5a | A-Z/Z | 5.1 | 10.2 | 15.3 | −1.8 | −17.1 | 0.49 |
| 2 | 2a | A-E | 5.5 | 9.1 | 14.6 | 3.2 | −11.4 | 0.44 |
| 3 | 2a | A-Z | 6.2 | 8.8 | 15.0 | −0.05 | −15.0 | 0.46 |
| 4 | 2a | B-E | 5.6 | 8.6 | 14.2 | 3.3 | −11.0 | 0.41 |
| 5 | 2a | B-Z | 6.2 | 10.3 | 16.5 | 0.53 | −16.0 | 0.35 |
a) Energies are given in kcal/mol. b) Values of asynchronicity are given in angstrom. c) Reference 7.
We think there are two major factors that potentially explain the energy gap between TS-II-A-Z (lowest-energy TS) and TS-II-B-Z (second lowest-energy TS) in the (3 + 2) cycloaddition of cyclic (E)-nitrone 2a (Supplementary Fig. S6). First, the stabilization effect associated with distortion in Mode A may influence the reduction of the energy not only in INT-III-A-Z but also in TS-II-A-Z. The other factor is the steric repulsion between the reactants and ligand. While replacing substituents in diamine (R,R)-3 from cyclohexyl groups to methyl groups reduced the value of ΔΔG‡ to 0.8 kcal/mol [TS-II-Me-A-Z: 17.7 kcal/mol vs. TS-II-Me-B-Z: 18.5 kcal/mol], (1R,2R,10bS)-4aa should be obtained as the major adduct. The trend in the geometrical distortion in TS-II-Mes using (R,R)-3c (R = Me) is also similar to that in the TS-IIs using (R,R)-3a (R = cHex). The Ni–O bond along the z-axis of TS-II-Me-A-Z is elongated to 2.11 Å, while that in TS-II-Me-B-Z is not elongated (Ni–O: 2.05 Å) (Supplementary Table S2, Supplementary Fig. S10).
Overall, our computational investigations suggest that the stereo-determining TS structure (TS-II-A-Z) in the developed (3 + 2) cycloaddition of 2a is determined integrally by (1) isomerism of the octahedral Ni(II) complex (Mode A vs. Mode B), (2) E/Z isomerism of the Ni(II)–enolate, and (3) steric repulsion between the reactants and ligand.