Experimental
GeneralMelting points were measured on a melting point apparatus (MP-500P; Yanaco Technical Science Co., Ltd., Tokyo, Japan) and were uncorrected. 1H-NMR spectra were obtained on a nuclear magnetic resonance spectrometer at 400 MHz (JNM-AL400 and JNM-ECZL400S; JEOL Ltd., Tokyo, Japan) using tetramethylsilane as an internal standard. IR spectra were recorded with an infrared spectrometer (HORIBA FT-720, HORIBA, Kyoto, Japan). Mass spectra were obtained with an electrospray ionization (ESI)-MS spectrometer (Expression CMS-L, Advion, Ithaca, U.S.A.) and ESI-time-of-flight (TOF)/MS (micrOTOF2-kp, Bruker, MA, U.S.A.). Column chromatography was performed on silica gel (Daisogel No.1001W; Daiso Co., Ltd., Osaka, Japan). Reactions were monitored by TLC (TLC silica gel 60F254, Merck KGaA, Darmstadt, Germany). The purities of the final compounds were assessed by HPLC (pump, LC-20AB; detector, SPD-20A; Shimadzu Corporation, Kyoto, Japan) using the COSMOSIL 5C18-AR-II column (5 µm, 4.6 × 150 mm; Nacalai Tesque, Kyoto, Japan).
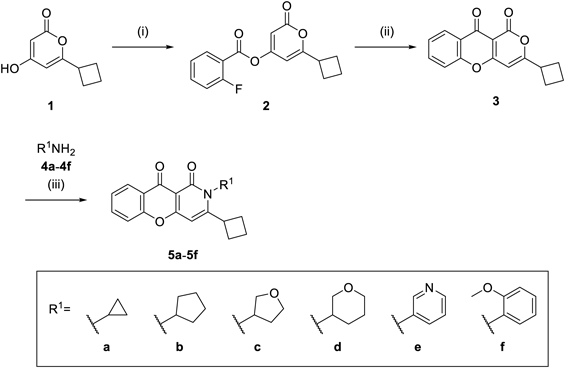
6-Cyclobutyl-2-oxo-2H-pyran-4-yl 2-Fluorobenzoate (2)Following the addition of Et3N (4.18 mL, 30.0 mmol) to a suspension of 1 (2.49 g, 15.0 mmol) in toluene (270 mL), 2-fluorobenzoyl chloride (2.68 mL, 22.5 mmol) was added dropwise at room temperature and stirred at the same temperature for 30 min. After the addition of saturated aqueous NH4Cl, the mixture was extracted with AcOEt. The organic layer was washed with saturated aqueous NH4Cl and saturated brine, and then dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 2 (4.8 g, quant.) as an oil. 1H-NMR (CDCl3) δ: 1.84–2.12 (2H, m), 2.22–2.40 (4H, m), 3.32–3.45 (1H, m), 6.04–6.09 (1H, m), 6.14–6.18 (1H, m), 7.16–7.36 (2H, m), 7.59–7.69 (1H, m), 7.97–8.08 (1H, m).
3-Cyclobutylpyrano[4,3-b]chromene-1,10-dione (3)Following the addition of KCN (1.47 g, 22.5 mmol), Et3N (3.14 mL, 22.5 mmol), and 18-crown-6 (0.40 g, 1.5 mmol) to a solution of 2 (4.80 g, 15.0 mmol) in toluene (375 mL), the mixture was stirred at room temperature for 10 min, at 60 °C for 30 min, and at 100 °C for 45 min. After cooling, water was added to the reaction mixture, which was then extracted with AcOEt. The organic layer was washed with saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 3 (1.08 g, 27% yield) as a solid. 1H-NMR (CDCl3) δ: 1.85–2.19 (2H, m), 2.27–2.46 (4H, m), 3.36–3.50 (1H, m), 6.23 (1H, s), 7.41–7.49 (2H, m), 7.66–7.73 (1H, m), 8.25–8.34 (1H, m).
3-Cyclobutyl-2-(tetrahydrofuran-3-yl)-2H-10-oxa-2-azaanthracene-1,9-dione (5d)Following the addition of Et3N 1.1 mL (8.0 mmol), 3 (270 mg, 1.00 mmol), and AcOH (1.4 mL) to a suspension of tetrahydropyran-3-ylamine hydrochloride (920 mg, 6.69 mmol) in CF3CH2OH (2.8 mL), the reaction mixture was stirred at 110 °C for 14 h. After cooling, water was added to the reaction mixture, which was then extracted with AcOEt. The organic layer was washed with saturated aqueous NaHCO3 and saturated brine and then dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography and t-BuOMe (3 mL) was added. The insoluble material was collected by filtration to give 5d (57 mg, 16% yield) as a solid. mp 194–196 °C; 1H-NMR (dimethyl sulfoxide (DMSO)-d6) δ: 1.62–2.00 (4H, m), 2.06–2.36 (3H, m), 2.38–2.52 (2H, m), 3.10–3.26 (1H, m), 3.40–3.64 (2H, m), 3.68–3.78 (1H, m), 3.86–3.97 (1H, m), 4.00–4.12 (1H, m), 4.79 (1H, t, J = 10.2 Hz), 6.13 (1H, s), 7.31–7.40 (2H, m), 7.58–7.77 (1H, m), 8.26–8.33 (1H, m); 13C-NMR (DMSO-d6) δ: 17.1 (s), 25.1 (s), 26.3 (s), 27.55 (s), 27.60 (s), 37.7 (s), 56.6 (s), 67.0 (s), 67.2 (s), 95.6 (s), 107.1 (s), 117.5 (s), 123.6 (s), 125.2 (s), 125.6 (s), 134.3 (s), 153.9 (s), 159.6 (s), 159.8 (s), 165.5 (s), 172.8 (s); IR (attenuated total reflectance (ATR)) cm−1: 1693. High resolution (HR)-MS (ESI-TOF) Calcd for C21H21NNaO4 (M + Na)+ 374.1368. Found 374.1394.
3-Cyclobutyl-2-cyclopropyl-2H-10-oxa-2-azaanthracene-1,9-dione (5a)Compound 5a was prepared from 3 according to the procedure for the synthesis of 5d. Yield was 27%. mp 276–281 °C; 1H-NMR (DMSO-d6) δ: 0.72–0.81 (2H, m), 1.12–1.22 (2H, m), 1.73–1.85 (1H, m), 1.95–2.11 (1H, m), 2.15–2.27 (2H, m), 2.35–2.45 (2H, m), 2.69–2.78 (1H, m), 3.95–4.08 (1H, m), 6.24 (1H, s), 7.41–7.49 (1H, m), 7.55 (1H, d, J = 8.3 Hz), 7.72–7.83 (1H, m), 8.05 (1H, d, J = 7.8 Hz); 13C-NMR (DMSO-d6) δ: 9.9 (s, 2C), 17.2 (s), 27.3 (s), 27.8 (s, 2C), 37.3 (s), 94.9 (s), 106.7 (s), 117.5 (s), 123.5 (s), 125.2 (s), 125.5 (s), 134.2 (s), 154.0 (s), 160.0 (s), 162.3 (s), 165.4 (s), 172.8 (s); IR (ATR) cm−1: 1685. HR-MS (ESI-TOF) Calcd for C19H17NNaO3 (M + Na)+ 330.1106. Found 330.1128.
3-Cyclobutyl-2-cyclopentyl-2H-10-oxa-2-azaanthracene-1,9-dione (5b)Compound 5b was prepared from 3 according to the procedure for the synthesis of 5d. Yield was 32%. mp 230–233 °C; 1H-NMR (DMSO-d6) δ: 1.47–1.64 (2H, m), 1.66–1.88 (3H, m), 1.90–2.08 (3H, m), 2.10–2.29 (4H, m), 2.30–2.43 (2H, m), 3.72–3.86 (1H, m), 4.43–4.60 (1H, m), 6.22 (1H, s), 7.37–7.49 (1H, m), 7.51–7.59 (1H, m), 7.72–7.83 (1H, m), 8.05 (1H, d, J = 7.8 Hz); 13C-NMR (DMSO-d6) δ: 17.1 (s), 25.7 (s, 2C), 27.7 (s, 2C), 28.1 (s, 2C), 37.7 (s), 58.4 (s), 94.9 (s), 107.0 (s), 117.5 (s), 123.6 (s), 125.1 (s), 125.5 (s), 134.2 (s), 153.9 (s), 158.8 (s), 159.8 (s), 165.3 (s), 173.0 (s); IR (ATR) cm−1: 1678. HR-MS (ESI-TOF) Calcd for C21H21NNaO3 (M + Na)+ 358.1419. Found 358.1428.
3-Cyclobutyl-2-(tetrahydrofuran-3-yl)-2H-10-oxa-2-azaanthracene-1,9-dione (5c)Compound 5c was prepared from 3 according to the procedure for the synthesis of 5d. Yield was 24%. mp 205–206 °C; 1H-NMR (DMSO-d6) δ: 1.70–2.45 (8H, m), 3.80–3.94 (4H, m), 3.99–4.07 (1H, m), 4.70–4.90 (1H, m), 6.29 (1H, s), 7.43–7.50 (1H, m), 7.53–7.61 (1H, m), 7.73–7.84 (1H, m), 8.06 (1H, d, J = 7.6 Hz); 13C-NMR (DMSO-d6) δ: 17.1 (s), 27.56 (s), 27.62 (s), 28.7 (s), 37.4 (s), 56.7 (s), 66.9 (s), 68.1 (s), 95.5 (s), 107.1 (s), 117.5 (s), 123.6 (s), 125.2 (s), 125.5 (s), 134.3 (s), 153.9 (s), 158.9 (s), 159.9 (s), 165.4 (s), 172.9 (s); IR (ATR) cm−1: 1682. HR-MS (ESI-TOF) Calcd for C20H20NO4 (M + H)+ 338.1392. Found 338.1372.
3-Cyclobutyl-2-pyridin-3-yl-2H-10-oxa-2-azaanthracene-1,9-dione (5e)Compound 5e was prepared from 3 according to the procedure for the synthesis of 5d. Yield was 32%. mp 219–220 °C; 1H-NMR (DMSO-d6) δ: 1.50–1.78 (4H, m), 2.00–2.20 (2H, m), 3.12–3.30 (1H, m), 6.48 (1H, s), 7.47–7.55 (1H, m), 7.57–7.67 (2H, m), 7.78–7.90 (2H, m), 8.04–8.13 (1H, m), 8.52 (1H, d, J = 2.0 Hz), 8.69 (1H, d, J = 4.4 Hz); 13C-NMR (DMSO-d6) δ: 16.7 (s), 26.9 (s), 27.0 (s), 37.7 (s), 95.4 (s), 106.9 (s), 117.7 (s), 123.5 (s), 124.1 (s), 125.4 (s), 125.6 (s), 134.2 (s), 134.5 (s), 136.8 (s), 149.5 (s), 149.6 (s), 154.0 (s), 159.3 (s), 159.4 (s), 166.4 (s), 172.7 (s); IR (ATR) cm−1: 1689. HR-MS (ESI-TOF) Calcd for C21H16N2NaO3 (M + Na)+ 367.1059. Found 367.1069.
3-Cyclobutyl-2-(2-methoxyphenyl)-2H-10-oxa-2-azaanthracene-1,9-dione (5f)Compound 5f was prepared from 3 according to the procedure for the synthesis of 5d. Yield was 29%. mp 248–258 °C; 1H-NMR (DMSO-d6) δ: 1.50–1.81 (4H, m), 1.95–2.22 (2H, m), 3.02–3.15 (1H, m), 3.72 (3H, s), 6.43 (1H, s), 7.07 (1H, t, J = 7.7 Hz), 7.16–7.27 (2H, m), 7.42–7.53 (2H, m), 7.60 (1H, d, J = 8.3 Hz), 7.75–7.86 (1H, m), 8.06 (1H, d, J = 7.4 Hz); 13C-NMR (DMSO-d6) δ: 16.8 (s), 26.6 (s), 27.2 (s), 37.6 (s), 55.5 (s), 94.8 (s), 106.8 (s), 112.1 (s), 117.6 (s), 120.5 (s), 123.6 (s), 125.3 (s), 125.5 (s), 125.7 (s), 129.8 (s), 130.5 (s), 134.4 (s), 154.0 (s), 154.5 (s), 158.6 (s), 160.0 (s), 166.4 (s), 172.8 (s); IR (ATR) cm−1: 1697. HR-MS (ESI-TOF) Calcd for C23H19NNaO4 (M + Na)+ 396.1212. Found 396.1213.
6-Cyclobutyl-4-hydroxy-1-phenylpyridine-2(1H)-one (6a)1 (1.86 g, 11.2 mmol) was suspended in AcOH (40 mL) and water (80 mL). Following the addition of aniline (1.03 mL, 11.2 mmol) to the reaction mixture, it was stirred at 85 °C for 18 h. After cooling, the reaction mixture was concentrated under reduced pressure. Following the addition of toluene (10 mL), the mixture was stirred at 50 °C for 10 min. The precipitate was collected by filtration. The resulting powder was washed with toluene and Et2O to give 6a (1.11 g, 41% yield) as a solid. 1H-NMR (CDCl3) δ: 1.45–1.65 (4H, m), 1.81–2.00 (2H, m), 2.94–3.09 (1H, m), 5.54 (1H, s), 5.91 (1H, s), 7.11–7.23 (2H, m), 7.34–7.54 (3H, m), 10.45–10.80 (1H, br).
2-(6-Cyclobutyl-2-oxo-1-phenyl-1,2-dihydropyridine-4-yl) 2-Fluorobenzoate (14a)Et3N (0.47 mL, 3.4 mmol) and 2-fluorobenzoyl chloride (0.30 mL, 2.5 mmol) were added to a suspension of 6a (400 mg, 1.66 mmol) in toluene (40 mL), and the mixture was stirred at room temperature for 40 min. Saturated aqueous NH4Cl was added to the reaction mixture, which was then extracted with AcOEt, and the organic layer was washed with saturated aqueous NH4Cl, water, and saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 14a (651 mg, quant.) as an oil. 1H-NMR (CDCl3) δ: 1.71–1.76 (4H, m), 1.93–2.04 (2H, m), 3.10–3.20 (1H, m), 6.16 (1H, d, J = 2.1 Hz), 6.44 (1H, d, J = 2.1 Hz), 7.11–7.24 (3H, m), 7.27–7.33 (1H, m), 7.42–7.54 (3H, m), 7.60–7.67 (1H, m), 8.06–8.15 (1H, m).
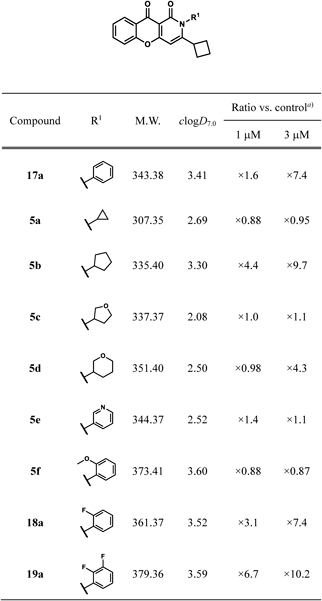
3-Cyclobutyl-2-phenyl-2H-10-oxa-2-azaanthracene-1,9-dione (17a)Following the addition of KCN (162 mg, 2.49 mmol), Et3N (0.35 mL, 2.5 mmol), and 18-crown-6 (44 mg, 0.17 mmol) to a solution of 14a (651 mg, 1.66 mmol) in toluene (40 mL), the mixture was stirred at room temperature for 16 h. After the addition of water, the mixture was extracted with AcOEt. The organic layer was washed with saturated brine and then dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography and t-BuOMe (10 mL) was added. The insoluble material was collected by filtration to give 17a (312 mg, 55% yield) as a solid. mp: 240–243 °C; 1H-NMR (DMSO-d6) δ: 1.56–1.73 (4H, m), 2.02–2.17 (2H, m), 3.11–3.23 (1H, m), 6.44 (1H, s), 7.25–7.33 (2H, m), 7.45–7.57 (4H, m), 7.61 (1H, d, J = 8.3 Hz), 7.77–7.84 (1H, m), 8.07 (1H, d, J = 7.6 Hz); 13C-NMR (DMSO-d6) δ: 16.9 (s), 27.3 (s, 2C), 38.0 (s), 94.9 (s), 107.0 (s), 117.7 (s), 123.6 (s), 125.4 (s), 125.6 (s), 128.7 (s), 128.9 (s, 2C), 129.2 (s, 2C), 134.5 (s), 137.4 (s), 154.1 (s), 159.3 (s), 159.6 (s), 166.3 (s), 172.8 (s); IR (ATR) cm−1: 1689; HR-MS (ESI-TOF) Calcd for C22H17NNaO3 (M + Na)+ 366.1106. Found 366.1122.
6-Cyclobutyl-1-(2-fluorophenyl)-4-hydroxy-1H-pyridin-2-one (7a)Compound 7a was prepared from 1 according to the procedure for the synthesis of 6a. Yield was 28%. 1H-NMR (CDCl3) δ: 1.61–1.81 (4H, m), 1.94–2.08 (2H, m), 3.01–3.13 (1H, m), 5.95–6.02 (2H, m), 7.14–7.26 (3H, m), 7.40–7.48 (1H, m).
2-[6-Cyclobutyl-1-(2-fluorophenyl)-2-oxo-1,2-dihydropyridine-4-yl] 2-Fluorobenzoate (15a)Compound 15a was prepared from 7a according to the procedure for the synthesis of 14a. Yield quant. 1H-NMR (CDCl3) δ: 1.62–1.85 (4H, m), 1.97–2.12 (2H, m), 3.09–3.21 (1H, m), 6.14–6.19 (1H, m), 6.43–6.47 (1H, m), 7.18–7.34 (5H, m), 7.42–7.50 (1H, m), 7.60–7.69 (1H, m), 8.05–8.12 (1H, m).
3-Cyclobutyl-2-(2-fluorophenyl)-2H-10-oxa-2-azaanthracene-1,9-dione (18a)Compound 18a was prepared from 15a according to the procedure for the synthesis of 17a. Yield was 24%. mp 220–249 °C; 1H-NMR (DMSO-d6) δ: 1.47–1.89 (4H, m), 1.95–2.09 (1H, m), 2.15–2.29 (1H, m), 3.11–3.25 (1H, m), 6.52 (1H, s), 7.33–7.54 (6H, m), 7.78–7.89 (1H, m), 8.06–8.12 (1H, m); 13C-NMR (DMSO-d6) δ: 16.8 (s), 26.81 (s), 26.84 (s), 37.6 (s), 95.6 (s), 106.8 (s), 116.2 (d), 117.7 (s), 123.6 (s), 124.8 (d), 125.2 (d), 125.5 (s), 125.6 (s), 130.8 (s), 131.4 (d), 134.5 (s), 154.0 (s), 157.4 (d), 158.5 (s), 159.3 (s), 166.6 (s), 172.7 (s); IR (ATR) cm−1: 1693; HR-MS (ESI-TOF) Calcd for C22H16FNNaO3 (M + Na)+ 384.1012. Found 384.1024.
6-Cyclobutyl-1-(2,3-difluorophenyl)-4-hydroxy-1H-pyridin-2-one (8a)Compound 8a was prepared from 1 according to the procedure for the synthesis of 6a. Yield was 27%. 1H-NMR (DMSO-d6) δ: 1.40–1.52 (1H, m), 1.53–2.10 (5H, m), 3.07 (1H, quintet, J = 8.6 Hz), 5.56 (1H, s), 5.88 (1H, s), 7.10–7.37 (2H, m), 7.50–7.60 (1H, m).
6-Cyclobutyl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 2-Fluorobenzoate (16a)Compound 16a was prepared from 8a according to the procedure for the synthesis of 14a. Yield quant. 1H-NMR (CDCl3) δ: 1.65–1.88 (4H, m), 1.99–2.15 (2H, m), 3.07–3.23 (1H, m), 6.18 (1H, d, J = 2.2 Hz), 6.46 (1H, d, J = 2.2 Hz), 6.98–7.07 (1H, m), 7.19–7.36 (4H, m), 7.60–7.70 (1H, m), 8.06–8.12 (1H, m).
3-Cyclobutyl-2-(2,3-difluorophenyl)-2H-10-oxa-2-azaanthracene-1,9-dione (19a)Compound 19a was prepared from 16a according to the procedure for the synthesis of 17a. Yield was 45%. mp 230–233 °C; 1H-NMR (DMSO-d6) δ: 1.50–1.95 (4H, m), 1.97–2.10 (1H, m), 2.18–2.30 (1H, m), 3.18–3.30 (1H, m), 6.55 (1H, s), 7.31–7.46 (2H, m), 7.47–7.55 (1H, m), 7.60–7.75 (2H, m), 7.78–7.89 (1H, m), 8.08 (1H, d, J = 7.6 Hz); 13C-NMR (DMSO-d6) δ: 16.8 (s), 26.8 (s), 26.9 (s), 37.4 (s), 96.0 (s), 106.8 (s), 117.7 (s), 118.7 (d), 123.5 (s), 125.0 (dd), 125.5 (s), 125.6 (s), 126.2 (d), 126.5 (d), 134.6 (s), 146.0 (dd), 149.9 (dd), 154.0 (s), 158.4 (s), 159.0 (s), 166.7 (s), 172.7 (s); IR (ATR) cm−1: 1697; HR-MS (ESI-TOF) Calcd for C22H15F2NNaO3 (M + Na)+ 402.0918. Found 402.0927.
4-Hydroxy-6-methyl-1-phenyl-1H-pyridin-2-one (6b)Compound 6b was prepared from 1b according to the procedure for the synthesis of 6a. Yield was 60%. 1H-NMR (DMSO-d6) δ: 1.82 (3H, s), 5.54 (1H, d, J = 3.0 Hz), 5.86–5.89 (1H, m), 7.16–7.21 (2H, m), 7.39–7.51 (3H, m), 10.51–10.64 (1H, br).
6-Methyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl 2-Fluorobenzoate (14b)Compound 14b was prepared from 6b according to the procedure for the synthesis of 14a. Yield was 98%. 1H-NMR (CDCl3) δ: 1.99 (3H, s), 6.15–6.18 (1H, m), 6.45 (1H, d, J = 2.2 Hz), 7.19–7.32 (4H, m), 7.44–7.56 (3H, m), 7.60–7.67 (1H, m), 8.07 (1H, td, J = 7.8, 1.7 Hz).
3-Methyl-2-phenyl-2H-10-oxa-2-azaanthracene-1,9-dione (17b)Compound 17b was prepared from 14b according to the procedure for the synthesis of 17a. Yield was 6.4%. mp 255–259 °C; 1H-NMR (DMSO-d6) δ: 2.02 (3H, s), 6.57 (1H, s), 7.30–7.35 (2H, m), 7.45–7.63 (5H, m), 7.77–7.83 (1H, m), 8.07 (1H, d, J = 7.8 Hz); 13C-NMR (DMSO-d6) δ: 21.8 (s), 97.6 (s), 107.0 (s), 117.7 (s), 123.6 (s), 125.4 (s), 125.6 (s), 128.4 (s, 2C), 128.8 (s), 129.5 (s, 2C), 134.4 (s), 137.9 (s), 153.98 (s), 154.01 (s), 159.3 (s), 166.0 (s), 172.9 (s); IR (ATR) cm−1: 1685; HR-MS (ESI-TOF) Calcd for C19H13NNaO3 (M + Na)+ 326.0793. Found 326.0794.
6-Butyl-4-hydroxy-1-phenyl-1H-pyridin-2-one (6c)Compound 6c was prepared from 1c according to the procedure for the synthesis of 6a. Yield was 33%. 1H-NMR (DMSO-d6) δ: 0.67 (3H, t, J = 7.3 Hz), 1.01–1.15 (2H, m), 1.21–1.34 (2H, m), 2.23 (2H, t, J = 7.6 Hz), 5.53 (1H, d, J = 2.4 Hz), 5.83 (1H, d, J = 2.4 Hz), 7.13–7.22 (2H, m), 7.37–7.53 (3H, m), 10.58 (1H, s).
6-Butyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl 2-Fluorobenzoate (14c)Compound 14c was prepared from 6c according to the procedure for the synthesis of 14a. Yield quant. 1H-NMR (DMSO-d6) δ: 0.77 (3H, t, J = 7.4 Hz), 1.13–1.30 (2H, m), 1.37–1.49 (2H, m), 2.23 (2H, t, J = 7.6 Hz), 6.15 (1H, d, J = 2.4 Hz), 6.44 (1H, d, J = 2.4 Hz), 7.18–7.34 (4H, m), 7.43–7.58 (3H, m), 7.60–7.69 (1H, m), 8.05–8.15 (1H, m).
3-Butyl-2-phenyl-2H-10-oxa-2-azaanthracene-1,9-dione (17c)Compound 17c was prepared from 14c according to the procedure for the synthesis of 17a. Yield was 17%. mp 244–247 °C; 1H-NMR (DMSO-d6) δ: 0.70 (3H, t, J = 7.3 Hz), 1.07–1.20 (2H, m), 1.35–1.47 (2H, m), 2.27 (2H, t, J = 8.6 Hz), 6.50 (1H, s), 7.30–7.38 (2H, m), 7.44–7.66 (5H, m), 7.76–7.85 (1H, m), 8.06 (1H, d, J = 8.0 Hz); 13C-NMR (DMSO-d6) δ: 13.3 (s), 21.5 (s), 29.1 (s), 33.2 (s), 96.7 (s), 107.0 (s), 117.7 (s), 123.6 (s), 125.4 (s), 125.6 (s), 128.80 (s, 2C), 128.83 (s), 129.4 (s, 2C), 134.5 (s), 137.5 (s), 154.1 (s), 157.5 (s), 159.4 (s), 166.1 (s), 172.9 (s); IR (ATR) cm−1: 1685; HR-MS (ESI-TOF) Calcd for C22H19NNaO3 (M + Na)+ 368.1263. Found 368.1272.
4-Hydroxy-6-isopropyl-1-phenyl-1H-pyridin-2-one (6d)Compound 6d was prepared from 1d according to the procedure for the synthesis of 6a. Yield was 32%. 1H-NMR (CDCl3) δ: 1.04 (6H, d, J = 6.8 Hz), 2.46 (1H, septet, J = 6.8 Hz), 5.97–6.03 (2H, m), 7.61–7.22 (2H, m), 7.41–7.54 (3H, m).
6-Isopropyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl 2-Fluorobenzoate (14d)Compound 14d was prepared from 6d according to the procedure for the synthesis of 14a. Yield was 95%. 1H-NMR (DMSO-d6) δ: 1.11 (6H, d, J = 6.7 Hz), 2.57 (1H, septet, J = 6.7 Hz), 6.17 (1H, d, J = 2.3 Hz), 6.43 (1H, d, J = 2.3 Hz), 7.19–7.33 (3H, m), 7.43–7.57 (4H, m), 7.60–7.68 (1H, m), 8.05–8.13 (1H, m).
3-Isopropyl-2-phenyl-2H-10-oxa-2-azaanthracene-1,9-dione (17d)Compound 17d was prepared from 14d according to the procedure for the synthesis of 17a. Yield was 5.1%. mp 209–211 °C; 1H-NMR (DMSO-d6) δ: 1.13 (6H, d, J = 6.6 Hz), 2.43 (1H, septet, J = 6.6 Hz), 6.55 (1H, s), 7.32–7.39 (2H, m), 7.45–7.64 (5H, m), 7.76–7.85 (1H, m), 8.07 (1H, d, J = 7.6 Hz); 13C-NMR (DMSO-d6) δ: 21.5 (s, 2C), 31.1 (s), 94.0 (s), 106.8 (s), 117.6 (s), 123.5 (s), 125.3 (s), 125.6 (s), 128.7 (s, 2C), 128.8 (s), 129.4 (s, 2C), 134.4 (s), 137.5 (s), 154.0 (s), 159.3 (s), 163.6 (s), 166.4 (s), 172.7 (s); IR (ATR) cm−1: 1693; HR-MS (ESI-TOF) Calcd for C21H17NNaO3 (M + Na)+ 354.1106. Found 354.1118.
7-(2,3-Difluorophenylamino)-2,2-dimethylpyrano-[4,3-d][1,3]dioxine-4,5-dione (10)Following the addition of 2,3-difluoroaniline (2.9 mL, 29 mmol) to a solution of 9 (3.00 g, 13.0 mmol) in CHCl3 (30 mL), the reaction mixture was stirred at room temperature for 1 h. The solvent was removed under reduced pressure, and water (40 mL) and t-BuOMe (40 mL) were added. The insoluble material was collected by filtration to give 10 (3.47 g, 83% yield) as a solid. 1H-NMR (DMSO-d6) δ: 1.67 (6H, s), 5.36 (1H, s), 7.24–7.49 (3H, m), 10.93–11.07 (1H, br).
6-(2,3-Difluorophenyl)-7-hydroxy-2,2-dimethyl-6H-[1,3]dioxino[5,4-c]pyridine-4,5-dione (11)Following the addition of NaH (p = 60%) (1.72 g, 43 mmol) to phenol (20 mL) under ice-cooling, the mixture was stirred at room temperature for 10 min. After the addition of 10 (3.47 g, 10.8 mmol) in phenol (5 mL) to the mixture, the reaction mixture was stirred at 110 °C for 15 min. After cooling, water (120 mL) was added and the reaction mixture was washed with Et2O. The separated aqueous layer was acidified by adding 6.0 M aqueous HCl solution (20 mL), and the mixture was extracted with AcOEt. The organic layer was washed with saturated brine and then dried over Na2SO4. The solvent was removed under reduced pressure and AcOEt (10 mL) and t-BuOMe (30 mL) was added. The insoluble material was collected by filtration to give 11 (1.13 g, 33% yield) as a solid. 1H-NMR (DMSO-d6) δ: 1.63 (6H, s), 5.09 (1H, s), 7.07–7.18 (1H, m), 7.20–7.30 (1H, m), 7.40–7.58 (1H, m).
6-(2,3-Difluorophenyl)-7-ethylamino-2,2-dimethyl-6H-[1,3]dioxino[5,4-c]pyridine-4,5-dione (12g)A suspension of 11 (2.04 g, 6.31 mmol) in POCl3 (17 mL) was stirred at 100 °C for 15 min. After cooling, the reaction mixture was concentrated under reduced pressure and saturated aqueous NaHCO3 was added. The mixture was extracted with AcOEt. The organic layer was washed with saturated aqueous NaHCO3 and saturated brine and then dried over Na2SO4. The solvent was removed under reduced pressure to give an intermediate (1.54 g).
The solid obtained was dissolved in CH2Cl2 (15 mL), and i-Pr2NEt (1.09 mL, 6.31 mmol) and 2.0 M EtNH2 in tetrahydrofuran (THF) (2.5 mL, 5.0 mmol) were added under ice-cooling. The reaction mixture was stirred at room temperature for 2.5 h. Water was added and the mixture was extracted with CHCl3. The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 12g (1.02 g, 46% yield) as an oil. 1H-NMR (CDCl3) δ: 1.15–1.23 (3H, m), 1.72–1.77 (6H, m), 3.14–3.26 (2H, m), 4.23–4.34 (1H, br), 5.16 (1H, s), 7.00–7.11 (1H, m), 7.26–7.42 (2H, m).
tert-Butyl [6-(2,3-Difluorophenyl)-2,2-dimethyl-4,5-dioxo-5,6-dihydro-4H-[1,3]dioxino[5,4-c]pyridin-7-yl]ethylcarbamate (12f)Following the addition of DMAP (52 mg, 0.43 mmol) and Boc2O (93 mg, 0.43 mmol) to a solution of 12g (100 mg, 0.285 mmol) in MeCN (1 mL), the reaction mixture was stirred at room temperature for 16 h. After the addition of Boc2O (93 mg, 0.43 mmol), the reaction mixture was stirred at 40 °C for 2 h and then at 60 °C for 3 h. Water was added and the mixture was extracted with AcOEt. The organic layer was washed with saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 12f (55 mg, 43% yield) as an oil. 1H-NMR (CDCl3) δ: 1.12–1.20 (3H, m), 1.37 (9H, s), 1.77–1.82 (6H, m), 2.80–3.10 (1H, br), 3.30–3.50 (1H, br), 5.92 (1H, s), 7.00–7.19 (2H, m), 7.26–7.34 (1H, m).
tert-Butyl [1-(2,3-Difluorophenyl)-4-hydroxy-6-oxo-1,6-dihydropyridin-2-yl]ethylcarbamate (8f)Following the addition of 4.0 M aqueous NaOH (0.40 mL, 1.6 mmol) to a solution of 12f (50 mg, 0.11 mmol) in 1,4-dioxane (1 mL), the reaction mixture was heated to reflux for 15 h. After cooling, 0.1 M aqueous HCl solution (30 mL) was added and the mixture was extracted with AcOEt. The organic layer was washed with saturated NaCl and dried over Na2SO4. The solvent was removed under reduced pressure to give 8f (32 mg, 79% yield) as a solid.
1H-NMR (CDCl3) δ: 1.09 (3H, t, J = 7.1 Hz), 1.38 (9H, s), 2.70–3.00 (1H, br), 3.25–3.45 (1H, br), 6.01 (1H, s), 6.08 (1H, s), 7.03–7.17 (2H, m), 7.24–7.33 (1H, m).
6-(tert-Butoxycarbonylethylamino)-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 2-Fluorobenzoate (16f)Compound 16f was prepared from 8f according to the procedure for the synthesis of 14a. Yield was 83%. 1H-NMR (CDCl3) δ: 1.13 (3H, t, J = 7.1 Hz), 1.40 (9H, s), 2.70–3.06 (1H, br), 3.20–3.52 (1H, br), 6.17–6.30 (1H, m), 6.55–6.65 (1H, m), 7.00–7.32 (5H, m), 7.60–7.69 (1H, m), 8.02–8.10 (1H, m).
tert-Butyl [2-(2,3-Difluorophenyl)-1,9-dioxo-2,9-dihydro-1H-10-oxa-2-azaanthracen-3-yl]ethylcarbamate (19f)Compound 19f was prepared from 16f according to the procedure for the synthesis of 17a. Yield was 30%. 1H-NMR (DMSO-d6) δ: 1.00–1.45 (12H, m), 3.20–3.60 (2H, m), 6.75–6.90 (1H, m), 7.27–7.46 (2H, m), 7.49–7.56 (1H, m), 7.57–7.75 (2H, m), 7.81–7.89 (1H, m), 8.07–8.13 (1H, m).
2-(2,3-Difluorophenyl)-3-ethylamino-2H-10-oxa-2-azaanthracene-1,9-dione (19g)After the addition of TFA (0.5 mL) to a solution of 19f (82 mg, 0.18 mmol) in CH2Cl2 (0.5 mL), the reaction mixture was stirred at room temperature for 1 h. Saturated aqueous NaHCO3 was added and the mixture was extracted with CHCl3. The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography and t-BuOMe (5 mL) was added. The insoluble material was collected by filtration to give 19g (47 mg, 73% yield) as a solid. mp 305–310 °C; 1H-NMR (DMSO-d6) δ: 1.09 (3H, t, J = 7.1 Hz), 3.20–3.30 (2H, m), 5.71 (1H, s), 6.80–6.88 (1H, m), 7.21–7.30 (1H, m), 7.34–7.48 (3H, m), 7.61–7.75 (2H, m), 7.99 (1H, dd, J = 7.6, 1.5 Hz); 13C-NMR (DMSO-d6) δ: 13.8 (s), 37.2 (s), 75.2 (s), 98.8 (s), 116.9 (s), 119.0 (d), 123.3 (s), 123.9 (d), 124.6 (s), 125.5 (s), 125.7 (dd), 126.9 (d), 133.6 (s), 147.0 (dd), 150.7 (dd), 153.5 (s), 154.1 (s), 158.2 (s), 167.7 (s), 171.6 (s); IR (ATR) cm−1: 1685; HR-MS (ESI-TOF) Calcd for C20H14F2N2NaO3 (M + Na)+ 391.0870. Found 391.0900.
7-Azetidin-1-yl-6-(2,3-difluorophenyl)-2,2-dimethyl-6H-[1,3]dioxino[5,4-c]pyridine-4,5-dione (12e)Compound 12e was prepared from 11 according to the procedure for the synthesis of 12g. Yield was 42%. 1H-NMR (CDCl3) δ: 1.70–1.75 (6H, m), 2.20 (2H, quintet, J = 7.8 Hz), 3.55–3.75 (4H, m), 4.88 (1H, s), 7.02–7.12 (1H, m), 7.14–7.35 (2H, m).
6-Azetidin-1-yl-1-(2,3-difluoro-phenyl)-4-hydroxy-1H-pyridin-2-one (8e)Compound 8e was prepared from 12e according to the procedure for the synthesis of 8f. Yield was 70%. 1H-NMR (DMSO-d6) δ: 1.99 (2H, quintet, J = 7.6 Hz), 3.34–3.44 (4H, m), 4.96 (1H, d, J = 2.2 Hz), 5.14 (1H, d, J = 2.2 Hz), 7.14–7.22 (1H, m), 7.26–7.36 (1H, m), 7.50–7.60 (1H, m), 10.45 (1H, s).
6-Azetidin-1-yl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 2-Fluorobenzate (16e)Compound 16e was prepared from 8e according to the procedure for the synthesis of 14a. Yield was 81%. 1H-NMR (CDCl3) δ: 2.12 (2H, quintet, J = 7.6 Hz), 3.47–3.63 (4H, m), 5.26 (1H, d, J = 2.2 Hz), 5.94 (1H, d, J = 2.2 Hz), 7.08–7.15 (1H, m), 7.17–7.34 (4H, m), 7.58–7.66 (1H, m), 8.01–8.11 (1H, m).
3-Azetidin-1-yl-2-(2,3-difluorophenyl)-2H-10-oxa-2-azaanthracene-1,9-dione (19e)Compound 19e was prepared from 16e according to the procedure for the synthesis of 17a. Yield was 3.8%. 1H-NMR (DMSO-d6) δ: 2.02–2.18 (2H, m), 3.54–3.73 (4H, m), 5.43 (1H, s), 7.32–7.50 (4H, m), 7.56–7.75 (2H, m), 7.94–8.04 (1H, m); 13C-NMR (DMSO-d6) δ: 15.0 (s), 53.1 (s, 2C), 76.8 (s), 98.7 (s), 116.9 (s), 119.0 (d), 123.2 (s), 124.6 (s), 124.8 (dd), 125.2 (d), 125.5 (s), 127.4 (d), 133.7 (s), 147.1 (dd), 149.9 (dd), 154.0 (s), 154.2 (s), 157.9 (s), 166.8 (s), 171.5 (s); IR (ATR) cm−1: 1682; HR-MS (ESI-TOF) Calcd for C21H14F2N2NaO3 (M + Na)+ 403.0870. Found 403.0905.
6-(2,3-Difluorophenyl)-7-methoxy-2,2-dimethyl-6H-[1,3]dioxino[5,4-c]pyridine-4,5-dione (13)Following the addition of i-Pr2NEt (0.770 mL, 4.49 mmol) and trimethyloxonium tetrafluoroborate (697 mg, 4.71 mmol) to a suspension of 11 (1.45 g, 4.49 mmol) in CH2Cl2 (15 mL), the reaction mixture was stirred at room temperature for 15 h under a N2 atmosphere. After the addition of i-Pr2NEt (0.390 mL, 2.28 mmol) and trimethyloxonium tetrafluoroborate (350 mg, 2.28 mmol), the reaction mixture was stirred for 5 h. Following the addition of 1.0 M aqueous HCl solution, the mixture was extracted with CHCl3. The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 13 (860 mg, 57% yield) as a solid. 1H-NMR (CDCl3) δ: 1.78 (6H, s), 3.88 (3H, s), 6.21 (1H, s), 6.98–7.07 (1H, m), 7.12–7.20 (1H, m), 7.22–7.32 (1H, m).
1-(2,3-Difluorophenyl)-4-hydroxy-6-methoxy-1H-pyridin-2-one (8h)Compound 8h was prepared from 13 according to the procedure for the synthesis of 8f. Yield was 8.4%. 1H-NMR (CDCl3) δ: 3.73 (3H, s) 5.37 (1H, s), 5.47 (1H, s), 7.10–7.22 (1H, m), 7.23–7.35 (1H, m), 7.45–7.58 (1H, m), 10.88 (1H, s).
1-(2,3-Difluorophenyl)-6-methoxy-2-oxo-1,2-dihydropyridin-4-yl 2-Fluorobenzoate (16h)Compound 16h was prepared from 8h according to the procedure for the synthesis of 14a. Yield was 83%. 1H-NMR (CDCl3) δ: 3.81 (3H, s), 5.65 (1H, d, J = 2.2 Hz), 6.23 (1H, d, J = 2.2 Hz), 7.03–7.10 (1H, m), 7.15–7.34 (4H, m), 7.60–7.68 (1H, m), 8.03–8.11 (1H, m).
2-(2,3-Difluorophenyl)-3-methoxy-2H-10-oxa-2-azaanthracene-1,9-dione (19h)Compound 19h was prepared from 16h according to the procedure for the synthesis of 17a. Yield was 18%. mp: 246–254 °C; 1H-NMR (DMSO-d6) δ: 3.97 (3H, s), 7.30–7.43 (2H, m), 7.54–7.53 (1H, m), 7.54–7.67 (2H, m), 7.75–7.84 (1H, m), 8.02–8.11 (1H, m); 13C-NMR (DMSO-d6) δ: 58.5 (s), 78.3 (s), 103.4 (s), 117.5 (s), 118.4 (d), 123.3 (s), 124.0 (d), 124.9 (dd), 125.5 (s), 125.6 (s), 126.5 (d), 134.4 (s), 145.9 (dd), 150.0 (dd), 154.1 (s), 157.3 (s), 160.4 (s), 168.7 (s), 172.2 (s); IR (ATR) cm−1: HR-MS (ESI-TOF) Calcd for C19H11F2NNaO4 (M + Na)+ 378.0554. Found 378.0581.
2-(2,3-Difluorophenyl)-3-hydroxy-2H-10-oxa-2-azaanthracene-1,9-dione (19i)After the addition of 5.1 M HBr in AcOH (1.3 mL, 6.6 mmol) to 19h (50 mg, 0.141 mmol), the reaction mixture was stirred at 80 °C for 1 h. After cooling, saturated aqueous NaHCO3 and NaCl were added and the mixture was extracted with AcOEt. The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography and t-BuOMe was added. The insoluble material was collected by filtration to give 19i (24 mg, 50% yield) as a solid. mp: 336–341 °C; 1H-NMR (DMSO-d6) δ: 5.10–5.51 (1H, m), 6.60–7.98 (8H, m); 13C-NMR (DMSO-d6) δ: 85.6 (s), 116.7 (s), 120.6 (d), 123.5 (s), 124.1 (s), 125.2 (s), 126.0 (dd), 128.8 (d), 131.5 (d), 132.1 (s), 134.6 (s), 147.9 (dd), 149.9 (dd), 154.6 (s), 162.1 (s), 164.2 (s), 165.0 (s), 174.0 (s). HR-MS (ESI-TOF) Calcd for C18H8F2NO4 (M − H)− 340.0421. Found 340.0425.
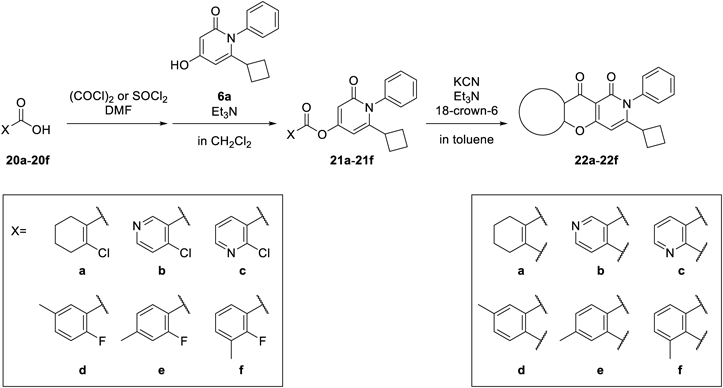
6-Cyclobutyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl 2-Chlorocyclohexenecarboxylate (21a)Following the addition of (COCl)2 (0.25 mL, 2.9 mmol) and N,N-dimethylformamide (DMF) 1 drop to a solution of 20a (300 mg, 1.87 mmol) under ice-cooling, the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was concentrated under reduced pressure to give an intermediate (0.40 g) as an oil.
Following the addition of Et3N (0.35 mL, 2.5 mmol) and the intermediate (0.40 g) to 6a (300 mg, 1.24 mmol) in CH2Cl2 (5 mL) under ice-cooling, the reaction mixture was stirred at room temperature for 1 h. After the removal of the solvent under reduced pressure, water was added and the mixture was extracted with AcOEt. The organic layer was washed with saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 21a (398 mg, 84% yield) as an oil. 1H-NMR (CDCl3) δ: 1.61–1.84 (8H, m), 1.95–2.03 (2H, m), 2.50–2.57 (4H, m), 3.06–3.17 (1H, m), 6.07 (1H, d, J = 2.4 Hz), 6.34 (1H, d, J = 2.4 Hz), 7.13–7.18 (2H, m), 7.40–7.51 (3H, m).
3-Cyclobutyl-2-phenyl-5,6,7,8-tetrahydro-2H-10-oxa-2-azaanthracene-1,9-dione (22a)Compound 22a was prepared from 21a according to the procedure for the synthesis of 17a. Yield was 4.8%. mp: 185–188 °C; 1H-NMR (DMSO-d6) δ: 1.54–1.69 (6H, m), 1.72–1.82 (2H, m), 1.97–2.10 (2H, m), 2.26–2.34 (2H, m), 2.57–2.63 (2H, m), 3.04–3.16 (1H, m), 6.32 (1H, s), 7.20–7.26 (2H, m), 7.44–7.54 (3H, m); 13C-NMR (DMSO-d6) δ: 16.9 (s), 20.6 (s), 20.9 (s), 21.2 (s), 26.6 (s), 27.3 (s, 2C), 37.8 (s), 94.6 (s), 108.4 (s), 121.2 (s), 128.6 (s), 128.9 (s, 2C), 129.1 (s, 2C), 137.6 (s), 157.5 (s), 159.3 (s), 160.8 (s), 165.6 (s), 174.0 (s); IR (ATR) cm−1: 1682; HR-MS (ESI-TOF) Calcd for C22H21NNaO3 (M + Na)+ 370.1419. Found 370.1425.
3-(6-Cyclobutyl-2-oxo-1-phenyl-1,2-dihydropyridine-4-yl) 4-Chloronicotininate (21b)Compound 21b was prepared from 20b according to the procedure for the synthesis of 21a. Yield was 79%. 1H-NMR (CDCl3) δ: 1.64–1.78 (4H, m), 1.93–2.04 (2H, m), 3.09–3.21 (1H, m), 6.16 (1H, d, J = 2.2 Hz), 6.47 (1H, d, J = 2.2 Hz), 7.16–7.22 (2H, m), 7.43–7.55 (4H, m), 8.69 (1H, d, J = 5.4 Hz), 9.24 (1H, s).
3-Cyclobutyl-2-phenyl-2H-10-oxa-2,7-diazaanthracene-1,9-dione (22b)Compound 22b was prepared from 21b according to the procedure for the synthesis of 17a. Yield was 25%. mp: 267–272 °C; 1H-NMR (DMSO-d6) δ: 1.57–1.75 (4H, m), 2.03–2.18 (2H, m), 3.13–3.24 (1H, m), 6.50 (1H, s), 7.26–7.32 (2H, m), 7.47–7.58 (3H, m), 7.63 (1H, d, J = 5.6 Hz), 8.84 (1H, d, J = 5.6 Hz), 9.17 (1H, s); 13C-NMR (DMSO-d6) δ: 16.9 (s), 27.4 (s, 2C), 38.1 (s), 94.9 (s), 108.3 (s), 112.5 (s), 119.3 (s), 128.7 (s, 2C), 128.9 (s), 129.3 (s, 2C), 137.2 (s), 148.8 (s), 153.9 (s), 158.9 (s), 159.5 (s), 160.7 (s), 166.6 (s), 172.2 (s); IR (ATR) cm−1: 1701; HR-MS (ESI-TOF) Calcd for C21H16N2NaO3 (M + Na)+ 367.1059. Found 367.1077.
6-Cyclobutyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl 2-Chloronicotinate (21c)Compound 21c was prepared from 20c according to the procedure for the synthesis of 21a. Yield was 89%. 1H-NMR (CDCl3) δ: 1.62–1.78 (4H, m), 1.92–2.09 (2H, m), 3.09–3.23 (1H, m), 6.19 (1H, d, J = 2.2 Hz), 6.50 (1H, d, J = 2.2 Hz), 7.16–7.22 (2H, m), 7.40–7.54 (4H, m), 8.43–8.40 (1H, m), 8.60–8.66 (1H, m).
7-Cyclobutyl-6-phenyl-6H-9-oxa-1,6-diazaanthracene-5,10-dione (22c)Compound 22c was prepared from 21c according to the procedure for the synthesis of 17a. Yield was 28%. mp: 215–216 °C; 1H-NMR (DMSO-d6) δ: 1.56–1.75 (4H, m), 2.05–2.21 (2H, m), 3.12–3.24 (1H, m), 6.57 (1H, s), 7.25–7.33 (2H, m), 7.46–7.58 (3H, m), 7.61 (1H, dd, J = 7.6, 4.6 Hz), 8.51 (1H, dd, J = 7.6, 1.8 Hz), 8.74 (1H, dd, J = 4.6, 1.8 Hz); 13C-NMR (DMSO-d6) δ: 16.8 (s), 27.3 (s, 2C), 38.0 (s), 95.1 (s), 106.9 (s), 118.5 (s), 122.6 (s), 128.7 (s, 2C), 128.8 (s), 129.2 (s, 2C), 136.4 (s), 137.2 (s), 153.1 (s), 158.7 (s), 159.1 (s), 160.2 (s), 166.4 (s), 173.4 (s); IR (ATR) cm−1: 1693; HR-MS (ESI-TOF) Calcd for C21H16N2NaO3 (M + Na)+ 367.1059. Found 367.1062.
(6-Cyclobutyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl) 2-Fluoro-5-methylbenzoate (21d)Compound 21d was prepared from 20d according to the procedure for the synthesis of 21a. Yield was 97%. 1H-NMR (CDCl3) δ: 1.63–1.77 (4H, m), 1.92–2.05 (2H, m), 2.41 (3H, s), 3.09–3.21 (1H, m), 6.15 (1H, d, J = 2.2 Hz), 6.43 (1H, d, J = 2.2 Hz), 7.07–7.15 (1H, m), 7.15–7.22 (2H, m), 7.38–7.54 (4H, m), 7.83–7.89 (1H, m).
3-Cyclobutyl-7-methyl-2-phenyl-2H-10-oxa-2-azaanthracene-1,9-dione (22d)Compound 22d was prepared from 13d according to the procedure for the synthesis of 21a. Yield was 8.5%. mp: 247–251 °C; 1H-NMR (DMSO-d6) δ: 1.53–1.73 (4H, m), 2.00–2.17 (2H, m), 2.43 (3H, s), 3.10–3.23 (1H, m), 6.43 (1H, s), 7.23–7.34 (2H, m), 7.44–7.57 (4H, m), 7.62 (1H, d, J = 8.0 Hz), 7.86 (1H, s); 13C-NMR (DMSO-d6) δ: 16.8 (s), 20.3 (s), 27.2 (s, 2C), 37.9 (s), 94.8 (s), 106.9 (s), 117.4 (s), 123.2 (s), 125.0 (s), 128.6 (s), 128.8 (s, 2C), 129.1 (s, 2C), 134.8 (s), 135.3 (s), 137.4 (s), 152.2 (s), 159.28 (s), 159.33 (s), 166.2 (s), 172.8 (s); IR (ATR) cm−1: 1689; HR-MS (ESI-TOF) Calcd for C23H19NNaO3 (M + Na)+ 380.1263. Found 380.1274.
6-Cyclobutyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl 2-Fluoro-4-methylbenzoate (21e)Compound 21e was prepared from 20e according to the procedure for the synthesis of 21a. Yield was 44%. 1H-NMR (CDCl3) δ: 1.62–1.75 (4H, m), 1.95–2.05 (2H, m), 2.45 (3H, s), 3.09–3.19 (1H, m), 6.16 (1H, d, J = 2.2 Hz), 6.43 (1H, d, J = 2.2 Hz), 7.04 (1H, d, J = 11.7 Hz), 7.09 (1H, d, J = 7.8 Hz), 7.16–7.21 (2H, m), 7.42–7.53 (3H, m), 7.97 (1H, t, J = 7.8 Hz).
3-Cyclobutyl-6-methyl-2-phenyl-2H-10-oxa-2-azaanthracene-1,9-dione (22e)Compound 22e was prepared from 21e according to the procedure for the synthesis of 17a. Yield was 13%. mp: 239–244 °C; 1H-NMR (DMSO-d6) δ: 1.50–1.72 (4H, m), 2.02–2.16 (2H, m), 2.48 (3H, s), 3.11–3.22 (1H, m), 6.41 (1H, s), 7.26–7.32 (3H, m), 7.42 (1H, s), 7.47–7.57 (3H, m), 7.95 (1H, d, J = 8.1 Hz); 13C-NMR (DMSO-d6) δ: 16.8 (s), 21.1 (s), 27.3 (s, 2C), 37.9 (s), 94.8 (s), 106.9 (s), 117.3 (s), 121.3 (s), 125.4 (s), 126.6 (s), 128.6 (s), 128.8 (s, 2C), 128.9 (s), 129.1 (s, 2C), 137.4 (s), 145.4 (s), 154.1 (s), 159.3 (s), 166.2 (s), 172.6 (s); IR (ATR) cm−1: 1689; HR-MS (ESI-TOF) Calcd for C23H19NNaO3 (M + Na)+ 380.1263. Found 380.1273.
(6-Cyclobutyl-2-oxo-1-phenyl-1,2-dihydropyridin-4-yl) 2-Fluoro-3-methylbenzoate (21f)Compound 21f was prepared from 20f according to the procedure for the synthesis of 21a. Yield was 80%. 1H-NMR (CDCl3) δ: 1.63–1.76 (4H, m), 1.92–2.06 (2H, m), 2.37 (3H, d, J = 2.0 Hz), 3.09–3.22 (1H, m), 6.16 (1H, d, J = 2.2 Hz), 6.43 (1H, d, J = 2.2 Hz), 7.12–7.21 (3H, m), 7.42–7.54 (4H, m), 7.86–7.96 (1H, m).
3-Cyclobutyl-5-methyl-2-phenyl-2H-10-oxa-2-azaanthracene-1,9-dione (22f)Compound 22f was prepared from 21f according to the procedure for the synthesis of 17a. Yield was 21%. mp: 239–242 °C; 1H-NMR (DMSO-d6) δ: 1.55–1.74 (4H, m), 2.05–2.19 (2H, m), 2.52 (3H, s), 3.12–3.23 (1H, m), 6.49 (1H, s), 7.24–7.32 (2H, m), 7.33–7.39 (1H, m), 7.45–7.58 (3H, m), 7.63–7.69 (1H, m), 7.85–7.93 (1H, m); 13C-NMR (DMSO-d6) δ: 14.9 (s), 16.8 (s), 27.3 (s, 2C), 38.0 (s), 95.0 (s), 106.7 (s), 123.1 (s), 123.5 (s), 124.7 (s), 126.7 (s), 128.7 (s), 128.8 (s, 2C), 129.1 (s, 2C), 135.1 (s), 137.4 (s), 152.4 (s), 159.29 (s), 159.31 (s), 166.0 (s), 173.0 (s); IR (ATR) cm−1: 1693; HR-MS (ESI-TOF) Calcd for C23H19NNaO3 (M + Na)+ 380.1263. Found 380.1268.
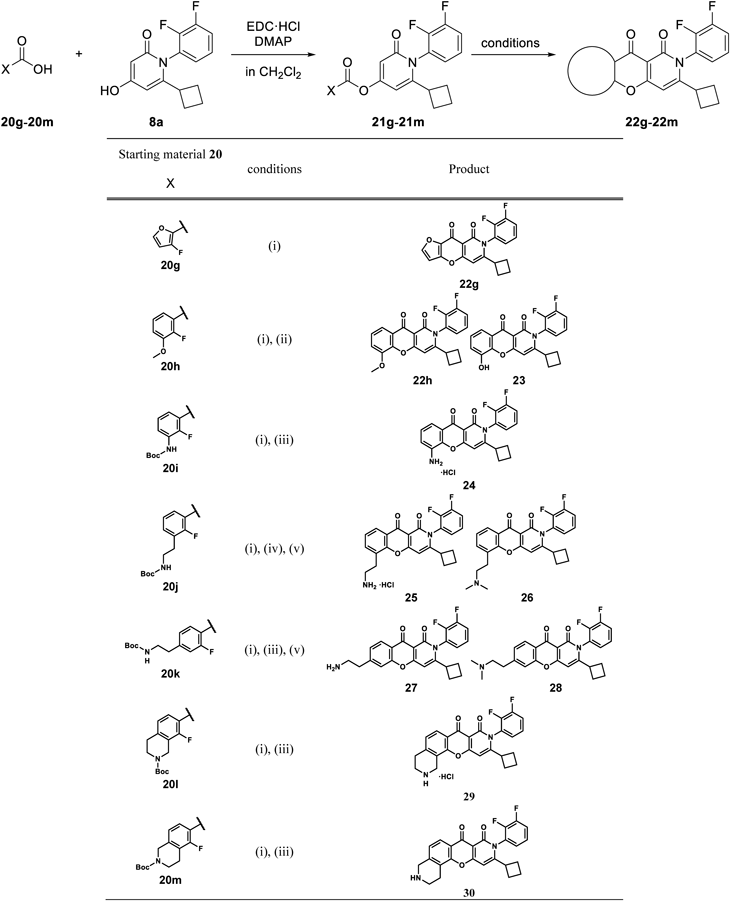
6-Cyclobutyl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 2-Fluoro-3-methoxy-benzoate (21h)Following the addition of EDC·HCl (114 mg, 0.595 mmol) and DMAP (7 mg, 0.05 mmol) to a suspension of 20h (92 mg, 0.54 mmol) and 8a (150 mg, 0.540 mmol) in CH2Cl2 (10 mL) and DMF (1 mL), the reaction mixture was stirred at room temperature for 1.5 h. After the addition of 20h (28 mg, 0.16 mmol) and EDC·HCl (34 mg, 0.18 mmol), the mixture was stirred for 0.5 h. Water was added and the mixture was extracted with AcOEt. The organic layer was washed with 1.0 M aqueous HCl solution, saturated aqueous NaHCO3, and saturated brine and then dried over Na2SO4. The solvent was removed under reduced pressure to give 21h (0.18 g, 77% yield) as an oil. 1H-NMR (CDCl3) δ: 1.65–1.89 (4H, m), 1.94–2.14 (2H, m), 3.08–3.20 (1H, m), 3.95 (3H, s), 6.15–6.21 (1H, m), 6.45–6.50 (1H, m), 6.98–7.05 (1H, m), 7.17–7.35 (4H, m), 7.56–7.65 (1H, m).
3-Cyclobutyl-2-(2,3-difluorophenyl)-5-methoxy-2H-10-oxa-2-azaanthracene-1,9-dione (22h)Compound 22h was prepared from 21h according to the procedure for the synthesis of 17a. Yield was 14%. mp: 217–218 °C; 1H-NMR (DMSO-d6) δ: 1.49–1.95 (4H, m), 1.96–2.12 (1H, m), 2.17–2.31 (1H, m), 3.17–3.28 (1H, m), 3.99 (3H, s), 6.53 (1H, s), 7.31–7.56 (4H, m), 7.57–7.74 (2H, m); 13C-NMR (DMSO-d6) δ: 16.9 (s), 26.9 (s), 27.0 (s), 37.5 (s), 56.3 (s), 96.2 (s), 106.8 (s), 116.0 (s), 116.1 (s), 118.7 (d), 124.6 (s), 125.1 (dd), 125.4 (s), 126.3 (d), 126.7 (d), 144.1 (s), 146.1 (dd), 148.1 (s), 150.0 (dd), 158.5 (s), 159.0 (s), 166.4 (s), 172.7 (s); IR (ATR) cm−1; 1697; HR-MS (ESI-TOF) Calcd for C23H17F2NNaO4 (M + Na)+ 432.1023. Found 432.1050.
3-Cyclobutyl-2-(2,3-difluorophenyl)-5-hydroxy-2H-10-oxa-2-azaanthracene-1,9-dione (23)Following the addition of 1.0 M BBr3 in CH2Cl2 (27 mL, 27 mmol) to a solution of 22h (1.1 g, 2.7 mmol) in CH2Cl2 (20 mL) under ice-cooling, the reaction mixture was stirred at room temperature for 2 h. Water (100 mL) was added dropwise under ice-cooling, followed by AcOEt (100 mL). After filtration of the insoluble material, the filtrate was dissolved in MeOH and dried over Na2SO4. The solvent was removed under reduced pressure and t-BuOMe was added. The insoluble material was collected by filtration to give 23 (346 mg, 33% yield) as a solid. mp: 298–301 °C; 1H-NMR (DMSO-d6) δ: 1.50–1.83 (4H, m), 1.84–1.94 (1H, m), 1.96–2.09 (1H, m), 3.19–3.32 (1H, m), 6.49 (1H, s), 7.24–7.30 (2H, m), 7.33–7.51 (3H, m), 7.61–7.71 (1H, m), 10.55 (1H, s); 13C-NMR (DMSO-d6) δ: 16.8 (s), 26.8 (s), 26.9 (s), 37.3 (s), 96.0 (s), 106.5 (s), 114.8 (s), 118.6 (d), 119.7 (s), 124.8 (s), 125.0 (dd), 125.3 (s), 126.2 (d), 126.6 (d), 143.4 (s), 146.0 (dd), 146.3 (s), 149.9 (dd), 158.5 (s), 158.7 (s), 166.2 (s), 172.9 (s); IR (ATR) cm−1: 1685; HR-MS (ESI-TOF) Calcd for C22H15F2NNaO4 (M + Na)+ 418.0867. Found 418.0905.
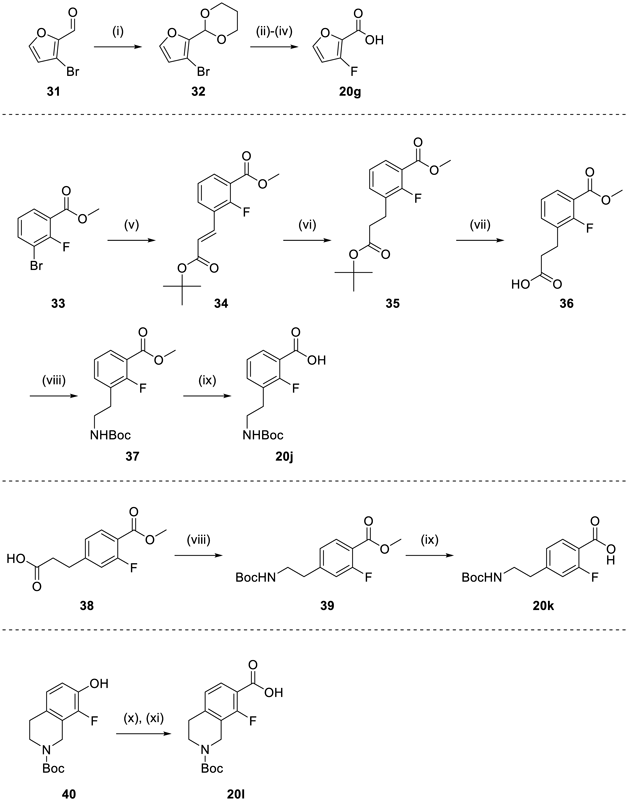
2-(3-Bromofuran-2-yl)-[1,3]dioxane (32)Following the addition of p-TsOH·H2O (1.66 g, 8.71 mmol) to a solution of 31 (3.81 g, 21.8 mmol) in propylene glycol (20 mL, 0.27 mol), the reaction mixture was stirred at room temperature for 2 h. After neutralization with 10% aqueous K2CO3, AcOEt was added. The organic layer was washed with water and dried over Na2SO4. The solvent was removed under reduced pressure to give 32 (4.70 g, 93% yield) as a solid. 1H-NMR (CDCl3) δ: 1.40–1.49 (1H, m), 2.21–2.38 (1H, m), 3.92–4.05 (2H, m), 4.18–4.35 (2H, m), 5.65 (1H, s), 6.41 (1H, d, J = 2.0 Hz), 7.37 (1H, d, J = 2.0 Hz).
3-Fluorofuran-2-carboxylic Acid (20g)After the addition of 1.6 M n-BuLi in n-hexane (13.2 mL, 21 mmol) to a solution of 32 (4.70 g, 20.2 mmol) in THF (20 mL) at −78 °C over 30 min, the reaction mixture was stirred at the same temperature for 15 min. Following the addition of N-fluorobenzenesulfonimide (6.69 g, 21.2 mmol) in THF (20 mL) over 45 min, the mixture was stirred at room temperature for 1 h. After the addition of water (18 mL) and Et3N (1.8 mL) under ice-cooling, Et2O was added. The mixture was washed with water and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give an intermediate (1.64 g) as an oil.
Following the addition of p-TsOH·H2O (0.72 g, 3.8 mmol) to a solution of the intermediate obtained (1.64 g) in 1,4-dioxane (8 mL) and water (4 mL), the reaction mixture was stirred at room temperature for 1 h. After neutralization with 10% aqueous K2CO3 solution, AcOEt was added. The mixture was washed with water and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give an intermediate (420 mg) as an oil.
Following the addition of 2-methyl-2-butene (3.90 mL, 36.8 mmol) to a solution of the intermediate obtained (420 mg) in t-BuOH (12 mL) and THF (6 mL), a solution of NaH2PO4 (3.53 g, 29.4 mmol) and NaClO2 (520 mg, 4.60 mmol) in water (4 mL) was added dropwise under ice-cooling and stirred at room temperature for 1.5 h. The mixture was extracted with AcOEt and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 20g (157 mg, 4.9% yield for 3 steps) as a solid. 1H-NMR (CDCl3) δ: 6.41–6.50 (1H, m), 7.41–7.48 (1H, m).
6-Cyclobutyl-7-(2,3-difluorophenyl)-7H-1,4-dioxa-7-azacyclopenta[b]naphthalene-8,9-dione (22g)Compound 22g was prepared from 20g according to the procedure for the synthesis of 22h. Yield was 2.3% for 2 steps. mp: 247–264 °C; 1H-NMR (DMSO-d6) δ: 1.47–1.95 (4H, m), 1.95–2.09 (1H, m), 2.15–2.29 (1H, m), 3.15–3.28 (1H, m), 6.60 (1H, s), 7.14 (1H, d, J = 2.0 Hz), 7.28–7.46 (2H, m), 7.58–7.72 (1H, m), 8.21 (1H, d, J = 2.0 Hz); 13C-NMR (DMSO-d6) δ: 16.7 (s), 26.7 (s), 26.8 (s), 37.1 (s), 96.1 (s), 103.0 (s), 109.7 (s), 118.7 (d), 125.0 (dd), 126.2 (d), 126.5 (d), 138.0 (s), 148.7 (dd), 148.8 (s), 149.9 (dd), 150.9 (s), 157.0 (s), 158.9 (s), 163.3 (s), 166.5 (s); IR (ATR) cm−1: 1689; HR-MS (ESI-TOF) Calcd for C20H13F2NNaO4 (M + Na)+ 392.0710. Found 390.0740.
6-Cyclobutyl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 3-tert-Butoxycarbonylamino-2-fluorobenzoate (21i)Compound 21i was prepared from 20i according to the procedure for the synthesis of 21h. Yield was 82%. 1H-NMR (CDCl3) δ: 1.45–1.63 (9H, m), 1.64–1.88 (4H, m), 1.98–2.13 (2H, m), 3.08–3.21 (1H, m), 6.14–6.20 (1H, m), 6.46 (1H, d, J = 2.2 Hz), 6.80–6.90 (1H, m), 6.98–7.04 (1H, m), 7.19–7.35 (3H, m), 7.65–7.71 (1H, m), 8.38–8.48 (1H, m).
tert-Butyl [3-Cyclobutyl-2-(2,3-difluorophenyl)-1,9-dioxo-2,9-dihydro-1H-10-oxa-2-azaanthracen-5-yl]carbamate (22i)Compound 22i was prepared from 21i according to the procedure for the synthesis of 17a. Yield was 32%. 1H-NMR (CDCl3) δ: 1.60 (9H, s), 1.70–1.94 (4H, m), 2.09–2.26 (2H, m), 3.14–3.28 (1H, m), 6.38 (1H, s), 6.97–7.04 (1H, m), 7.05–7.13 (1H, m), 7.18–7.38 (3H, m), 7.93 (1H, dd, J = 7.8, 1.2 Hz), 8.34–8.46 (1H, m).
5-Amino-3-cyclobutyl-2-(2,3-difluorophenyl)-2H-10-oxa-2-azaanthracene-1,9-dione Hydrochloride (24)After the addition of 4.0 M HCl in 1,4-dioxane (0.89 mL, 3.6 mmol) to 22i (117 mg, 0.237 mmol) under ice-cooling, the reaction mixture was stirred at the same temperature for 50 min and at room temperature for 1 h. Following the addition of 1,4-dioxane (2 mL) and AcOEt (1 mL), the insoluble material was collected by filtration to give 24 (84.8 mg, 83% yield) as a solid. mp: >152 °C (decomp.); 1H-NMR (DMSO-d6) δ: 1.53–2.25 (6H, m), 3.20–3.32 (1H, m), 6.60 (1H, s), 7.01–7.06 (1H, m), 7.11–7.21 (2H, m), 7.32–7.46 (2H, m), 7.62–7.72 (1H, m); 13C-NMR (DMSO-d6) δ: 16.8 (s), 26.8 (s), 26.9 (s), 37.3 (s), 96.1 (s), 106.3 (s), 111.6 (s), 117.8 (s), 118.6 (d), 123.9 (s), 125.0 (dd), 125.4 (s), 126.3 (d), 126.7 (d), 137.0 (s), 142.2 (s), 146.0 (dd), 149.9 (dd), 158.2 (s), 158.6 (s), 166.0 (s), 173.2 (s); IR (ATR) cm−1: 1689; HR-MS (ESI-TOF) Calcd for C22H16F2N2NaO3 (M + Na)+ 417.1027. Found 417.1068.
Methyl 4-(2-tert-Butoxycarbonylaminoethyl)-2-fluorobenzoate (39)Following the addition of Et3N (9.7 mL, 70 mmol) and diphenylphosphoryl azide (DPPA) (15 mL, 70 mmol) to a suspension of 38 (11.4 g, 46.6 mmol) in t-BuOH (94 mL), the reaction mixture was heated to 100 °C over 30 min and then stirred at the same temperature for 3.5 h. After cooling, water was added and the mixture was extracted with AcOEt. The organic layer was washed with saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 39 (6.61 g, 48% yield) as a solid. 1H-NMR (CDCl3) δ: 1.43 (9H, s), 2.55 (2H, t, J = 7.8 Hz), 3.30–3.46 (2H, m), 3.92 (3H, s), 4.41–4.60 (1H, br), 6.92–7.06 (2H, m), 7.82–7.90 (1H, m).
4-(2-tert-Butoxycarbonylaminoethyl)-2-fluorobenzoic Acid (20k)Following the addition of 1.0 M aqueous NaOH solution (67 mL, 67 mmol) to a solution of 39 (6.61 g, 22.2 mmol) in MeOH (130 mL), the reaction mixture was stirred at 40 °C for 1 h. After cooling, 1.0 M aqueous HCl solution (80 mL, 80 mmol) was added and the mixture was extracted with AcOEt. The organic layer was washed with saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure to give 20k (6.28 g, quant.) as a solid. 1H-NMR (CDCl3) δ: 1.34 (9H, s), 2.72–2.82 (2H, m), 3.12–3.23 (2H, m), 6.78–6.94 (1H, m), 7.03–7.23 (2H, m), 7.70–7.83 (1H, m), 12.84–13.19 (1H, br).
6-Cyclobutyl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 4-(2-tert-Butoxycarbonyl-aminoethyl)-2-fluorobenzoate (21k)Compound 21k was prepared from 20k according to the procedure for the synthesis of 21h. Yield was 99%. 1H-NMR (CDCl3) δ: 1.45 (9H, s), 1.64–1.93 (4H, m), 1.96–2.18 (2H, m), 2.81–2.98 (2H, m), 3.08–3.23 (1H, m), 3.35–3.52 (2H, m), 4.47–4.64 (1H, m), 6.15–6.20 (1H, m), 6.41–6.47 (1H, m), 6.97–7.17 (5H, m), 7.70–7.83 (1H, m).
tert-Butyl{2-[3-Cyclobutyl-2-(2,3-difluorophenyl)-1,9-dioxo-2,9-dihydro-1H-10-oxa-2-azaanthracen-6-yl]ethyl}carbamate (22k)Compound 22k was prepared from 21k according to the procedure for the synthesis of 17a. mp: 228–231 °C; 1H-NMR (DMSO-d6) δ: 1.11 (9H, s), 1.50–1.94 (4H, m), 1.97–2.10 (1H, m), 2.17–2.30 (1H, m), 2.87 (2H, t, J = 6.4 Hz), 3.18–3.30 (3H, m), 6.51 (1H, s), 6.90–6.99 (1H, br), 7.30–7.47 (4H, m), 7.60–7.72 (1H, m), 7.98 (1H, d, J = 8.1 Hz).
6-(2-Aminoethyl)-3-cyclobutyl-2-(2,3-difluorophenyl)-2H-10-oxa-2-azaanthracene-1,9-dione (27)Compound 27 was prepared from 20k according to the procedure for the synthesis of 24. 1H-NMR (DMSO-d6) δ: 1.72–1.93 (4H, m), 2.07–2.23 (2H, m), 2.89 (2H, t, J = 6.7 Hz), 3.07 (2H, t, J = 6.7 Hz), 3.13–3.26 (1H, m), 6.28 (1H, s), 6.98–7.07 (1H, m), 7.18–7.38 (4H, m), 8.23 (1H, d, J = 7.8 Hz).
3-Cyclobutyl-2-(2,3-difluorophenyl)-6-(2-dimethylaminoethyl)-2H-10-oxa-2-azaanthracene-1,9-dione (28)Following the addition of Et3N (0.030 mL, 0.22 mmol), formalin (p = 37%) (0.080 mL, 1.1 mmol), and NaBH(OAc)3 (228 mg, 1.08 mmol) to a solution of 27 (91 mg, 0.22 mmol) in MeOH (10 mL), the reaction mixture was stirred at room temperature for 1 h. The reaction mixture was concentrated under reduced pressure and saturated aqueous NaHCO3 was added. The mixture was extracted with AcOEt. The organic layer was washed with saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography and AcOEt (1 mL) was added. The insoluble material was collected by filtration to give 28 (40 mg, 41% yield) as a solid. mp: 174–177 °C; 1H-NMR (DMSO-d6) δ: 1.51–1.94 (4H, m), 1.97–2.09 (1H, m), 2.17–2.30 (1H, m), 2.34 (6H, s), 2.75 (2H, t, J = 7.0 Hz), 2.95 (2H, t, J = 7.0 Hz), 6.50 (1H, s), 7.31–7.47 (3H, m), 7.51 (1H, s), 7.60–7.71 (1H, m), 7.98 (1H, d, J = 8.1 Hz); 13C-NMR (DMSO-d6) δ: 16.8 (s), 26.8 (s), 26.9 (s), 32.8 (s), 37.3 (s), 44.9 (s, 2C), 59.6 (s), 96.0 (s), 106.7 (s), 117.2 (s), 118.6 (d), 121.6 (s), 125.0 (dd), 125.3 (s), 126.2 (d), 126.4 (s), 126.6 (d), 146.0 (dd), 148.6 (s), 149.9 (dd), 154.0 (s), 158.4 (s), 158.8 (s), 166.6 (s), 172.5 (s); IR (ATR) cm−1; HR-MS (ESI-TOF) Calcd for C26H24F2N2NaO3 (M + Na)+ 473.1653. Found 473.1683.
Methyl 3-(2-tert-Butoxycarbonylvinyl)-2-fluorobenzoate (34)After the addition of Pd(OAc)2 (449 mg, 2.00 mmol) and P(o-tolyl)3 (1.22 g, 4.00 mmol) to DMF (20 mL), the reaction mixture was stirred at room temperature for 5 min. Following the addition of 33 (4.66 g, 20.0 mmol), tert-butyl acrylate (5.8 mL, 40 mmol), and Et3N (5.8 mL, 40 mmol), the reaction mixture was stirred at 100 °C for 16 h and at 120 °C for 2 h. After cooling, AcOEt was added and the mixture was extracted with AcOEt. The organic layer was washed with water and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 34 (4.2 g, 68% yield) as a solid. 1H-NMR (CDCl3) δ: 1.54 (9H, s), 3.94 (3H, s), 6.47 (1H, d, J = 16.1 Hz), 7.21 (1H, t, J = 7.8 Hz), 7.64–7.80 (2H, m), 7.86–7.96 (1H, m).
Methyl 3-(2-tert-Butoxycarbonylethyl)-2-fluorobenzoate (35)A solution of 34 (4.24 g, 13.7 mmol) in MeOH (75 mL) was hydrogenated at 0.4 MPa in the presence of 10% Pd-C (770 mg) at room temperature for 15 h. After removal of the catalyst by filtration, the filtrate was evaporated under reduced pressure to give 35 (3.80 g, 98% yield) as an oil. 1H-NMR (CDCl3) δ: 1.40 (9H, s), 2.55 (2H, t, J = 7.6 Hz), 2.98 (2H, t, J = 7.6 Hz), 3.92 (3H, s), 7.07–7.14 (1H, m), 7.36–7.44 (1H, m), 7.72–7.80 (1H, m).
3-(2-Fluoro-3-methoxycarbonylphenyl)propionic Acid (36)After the addition of 35 (3.80 g, 13.5 mmol) to CH2Cl2 (13.5 mL) and TFA (13.5 mL), the reaction mixture was stirred at room temperature for 2.5 h. The reaction mixture was concentrated under reduced pressure and AcOEt was then added. The mixture was washed with water and dried over Na2SO4. The solvent was removed under reduced pressure to give 36 (3.67 g, 87% yield) as an oil. 1H-NMR (CDCl3) δ: 2.55 (2H, t, J = 7.6 Hz), 2.89 (2H, t, J = 7.6 Hz), 3.85 (3H, s), 7.19–7.28 (1H, m), 7.54–7.62 (1H, m), 7.64–7.78 (1H, m).
Methyl 3-(2-tert-Butoxycarbonylaminoethyl)-2-fluorobenzoate (37)Following the addition of Et3N (2.43 mL, 17.5 mmol) and DPPA (3.77 mL, 17.5 mmol) to a suspension of 36 (3.67 g, 11.7 mmol) in t-BuOH (25 mL), the reaction mixture was heated to 100 °C over 23 min, and the mixture was stirred at the same temperature for 1.5 h. After cooling, water was added and the mixture was extracted with AcOEt. The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 37 (1.41 g, 41% yield) as an oil. 1H-NMR (CDCl3) δ: 1.42 (9H, s), 2.82–2.97 (2H, m), 3.29–3.45 (2H, m), 3.92 (3H, s), 4.48–4.64 (1H, br), 7.13 (1H, t, J = 7.6 Hz), 7.32–7.44 (1H, m), 7.74–7.83 (1H, m).
3-(2-tert-Butoxycarbonylaminoethyl)-2-fluorobenzoic Acid (20j)Following the addition of 1.0 M aqueous NaOH (14.2 mL, 14 mmol) to a solution of 37 (1.41 g, 4.74 mmol) in MeOH (36 mL), the reaction mixture was stirred at 40 °C for 40 min. After cooling, 1.0 M aqueous HCl solution was added to the reaction solution to make it weakly acidic. The mixture was extracted with AcOEt and dried over Na2SO4. The solvent was removed under reduced pressure to give 20j (1.07 g, 80% yield) as a solid. 1H-NMR (DMSO-d6) δ: 1.34 (9H, s), 2.70–2.83 (2H, m), 3.05–3.20 (2H, m), 6.81–6.99 (1H, m), 7.19 (1H, t, J = 7.6 Hz), 7.35–7.53 (1H, m), 7.56–7.78 (1H, m).
6-Cyclobutyl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 3-(2-tert-Butoxycarbonylaminoethyl)-2-fluorobenzoate (21j)Compound 21j was prepared from 20j according to the procedure for the synthesis of 21h. Yield quant. 1H-NMR (CDCl3) δ: 1.43 (9H, s), 1.67–1.90 (4H, m), 1.97–2.16 (2H, m), 2.83–3.03 (2H, m), 3.05–3.26 (1H, m), 3.34–3.48 (2H, m), 4.52–4.67 (1H, m), 6.17 (1H, s), 6.46 (1H, s), 6.97–7.06 (1H, m), 7.15–7.36 (3H, m), 7.47–7.57 (1H, m), 7.91–7.99 (1H, m).
tert-Butyl{2-[3-Cyclobutyl-2-(2,3-difluorophenyl)-1,9-dioxo-2,9-dihydro-1H-10-oxa-2-azaanthracen-5-yl]ethyl}carbamate (22j)Compound 22j was prepared from 21j according to the procedure for the synthesis of 17a. mp: 264–265 °C; 1H-NMR (CDCl3) δ: 1.29 (9H, s), 1.51–2.11 (5H, m), 2.17–2.32 (1H, m), 2.94–3.09 (2H, m), 3.18–3.42 (3H, m), 6.70 (1H, s), 6.85–6.98 (1H, m), 7.30–7.49 (3H, m), 7.56–7.73 (2H, m), 7.93 (1H, d, J = 7.3 Hz) ; IR (ATR) cm−1: 1685.
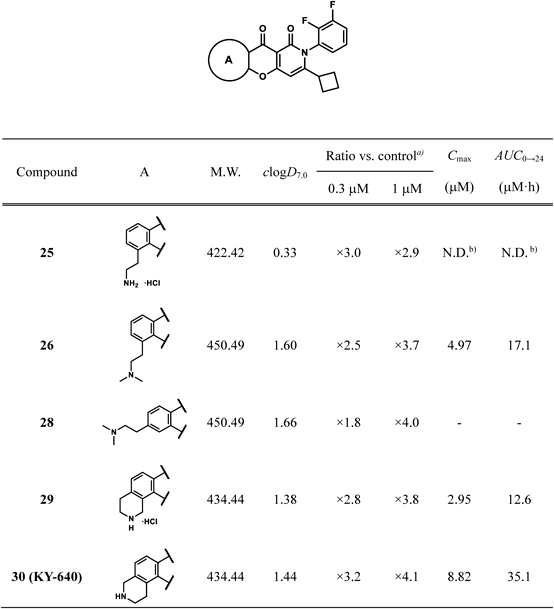
5-(2-Aminoethyl)-3-cyclobutyl-2-(2,3-difluorophenyl)-2H-10-oxa-2-azaanthracene-1,9-dione Hydrochloride (25)Following the addition of 9.1 M HCl in i-PrOH (0.28 mL, 2.6 mmol) to a suspension of 22j (134 mg, 0.256 mmol) in AcOEt (0.57 mL) under ice-cooling, the reaction mixture was stirred at the same temperature for 2 h. After the addition of 9.1 M HCl in i-PrOH (0.28 mL, 2.6 mmol) under ice-cooling, the mixture was stirred at the same temperature for 1 h. The insoluble material was collected by filtration to give 25 (51 mg, 47% yield) as a solid. mp: >242 °C (decomp.); 1H-NMR (DMSO-d6) δ: 1.52–1.98 (4H, m), 1.98–2.13 (1H, m), 2.19–2.34 (1H, m), 3.14–3.29 (5H, m), 6.81 (1H, s), 7.27–7.52 (3H, m), 7.60–7.79 (2H, m), 7.97 (1H, d, J = 7.6 Hz), 8.02–8.19 (3H, br); 13C-NMR (DMSO-d6) δ: 16.9 (s), 26.9 (s), 27.1 (s), 27.2 (s, 2C), 37.5 (s), 96.8 (s), 106.6 (s), 118.8 (d), 123.7 (s), 124.4 (s), 125.1 (s) 125.2 (d), 126.3 (d), 126.6 (s), 126.7 (s), 135.6 (s), 146.0 (dd), 150.0 (dd), 152.7 (s), 158.6 (s), 158.9 (s), 166.6 (s), 173.1 (s); IR (ATR) cm−1: 1689; HR-MS (ESI-TOF) Calcd for C24H20F2N2NaO3 (M + Na)+ 445.1340. Found 445.1368.
3-Cyclobutyl-2-(2,3-difluorophenyl)-5-(2-dimethylaminoethyl)-2H-10-oxa-2-azaanthracene-1,9-dione (26)Compound 26 was prepared from 25 according to the procedure for the synthesis of 28. mp: 191–194 °C; 1H-NMR (DMSO-d6) δ: 1.51–2.14 (5H, m), 2.16–2.31 (1H, m), 2.43 (6H, s), 2.74–2.93 (2H, m), 3.05–3.50 (3H, m), 6.58 (1H, s), 7.24–7.55 (3H, m), 7.57–7.82 (2H, m), 7.94 (1H, d, J = 7.6 Hz); 13C-NMR (DMSO-d6) δ: 16.8 (s), 26.5 (s), 26.9 (s), 27.0 (s), 37.4 (s), 45.1 (s, 2C), 59.1 (s), 96.2 (s), 106.5 (s), 118.7 (d), 123.4 (s), 123.6 (s), 125.0 (s), 125.1 (d), 126.3 (d), 126.7 (d), 129.8 (s), 135.0 (s), 146.1 (dd), 150.0 (dd), 152.3 (s), 158.5 (s), 159.0 (s), 166.5 (s), 173.0 (s); IR (ATR) cm−1: 1685; HR-MS (ESI-TOF) Calcd for C26H24F2N2NaO3 (M + Na)+ 473.1653. Found 473.1697.
2-tert-Butoxycarbonyl-8-fluoro-1,2,3,4-tetrahydroisoquinoline-7-carboxylic Acid (20l)Following the addition of pyridine (6.7 mL, 83 mmol) to a solution of 40 (4.44 mL, 16.6 mmol) in CH2Cl2 (60 mL), Tf2O (4.08 mL, 24.9 mmol) was added dropwise under ice-cooling, and the reaction mixture was stirred at room temperature for 1 h. Water was added and the mixture was extracted with CHCl3. The organic layer was washed with 1.0 M aqueous HCl solution and dried over Na2SO4. The solvent was removed under reduced pressure.
The residue obtained was dissolved in DMF (70 mL), and HCO2Li·H2O (3.50 g, 50.0 mmol), 9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (577 mg, 0.997 mmol), tris(dibenzylideneacetone)dipalladium (912 mg, 0.996 mmol), LiCl (2.53 g, 59.7 mmol), i-Pr2NEt (6.8 mL, 40 mmol), and Ac2O (3.8 mL, 40 mmol) were added. The reaction mixture was stirred at 80 °C for 14 h under a N2 atmosphere. After cooling, water was added to the mixture, and the insoluble material was filtered off. One molar aqueous HCl solution was added to the filtrate to acidify it, and the mixture was extracted with AcOEt. The organic layer was washed with water and dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography to give 20l (2.61 g, 83% yield) as a solid. 1H-NMR (CDCl3) δ: 1.50 (9H, s), 2.82–2.93 (2H, m), 3.57–3.76 (2H, m), 4.60–4.73 (2H, br), 7.02 (1H, d, J = 8.0 Hz), 7.78–7.86 (1H, m).
6-Cyclobutyl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl 2-tert-Butoxycarbonyl-8-fluoro-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (21l)Compound 21l was prepared from 20l according to the procedure for the synthesis of 21f. Yield was 71%. 1H-NMR (CDCl3) δ: 1.51 (9H, m), 1.64–1.89 (4H, m), 1.96–2.14 (2H, m), 2.84–2.98 (2H, m), 3.07–3.22 (1H, m), 3.61–3.76 (2H, m), 4.58–4.73 (2H, br), 6.14–6.20 (1H, m), 6.41–6.47 (1H, m), 6.95–7.35 (4H, m), 7.81–7.92 (1H, m).
tert-Butyl 10-Cyclobutyl-9-(2,3-difluorophenyl)-7,8-dioxo-4,7,8,9-tetrahydro-1H, 3H-12-oxa-2,9-diazabenzo[a]anthracene-2-carboxylate (22l)Compound 22l was prepared from 21l according to the procedure for the synthesis of 17a. Yield was 37%. 1H-NMR (CDCl3) δ: 1.47–1.68 (9H, m), 1.72–1.96 (4H, m), 2.07–2.26 (2H, m), 2.89–3.02 (2H, m), 3.14–3.29 (1H, m), 3.62–3.83 (2H, m), 4.71–4.95 (2H, m), 6.32 (1H, s), 6.97–7.06 (1H, m), 7.16–7.36 (3H, m), 8.11 (1H, d, J = 8.3 Hz).
10-Cyclobutyl-9-(2,3-difluorophenyl)-1,2,3,4-tetrahydro-9H-12-oxa-2,9-diazabenzo[a]anthracene-7,8-dione Hydrochloride (29)Compound 29 was prepared from 22l according to the procedure for the synthesis of 24. Yield was 85%. mp: 235–243 °C; 1H-NMR (DMSO-d6) δ: 1.51–2.12 (5H, m), 2.16–2.31 (1H, m), 3.13–3.20 (2H, m), 3.21–3.31 (1H, m), 3.43–3.53 (2H, m), 4.43–4.60 (2H, m), 6.68 (1H, s), 7.25–7.48 (3H, m), 7.58–7.74 (3H, m), 7.96 (1H, d, J = 8.0 Hz), 9.51–9.88 (2H, br); 13C-NMR (DMSO-d6) δ: 16.8 (s), 18.5 (s), 25.0 (s), 26.9 (s), 27.0 (s), 37.5 (s), 56.0 (s), 96.3 (s), 106.9 (s), 118.4 (s), 118.8 (d), 121.7 (s), 124.0 (s), 125.1 (dd), 125.9 (s), 126.3 (d), 126.6 (d), 139.5 (s), 146.0 (dd), 150.0 (dd), 150.8 (s), 158.5 (s), 159.2 (s), 166.4 (s), 172.4 (s); IR (ATR) cm−1: 1689; HR-MS (ESI-TOF) Calcd for C25H20F2N2NaO3 (M + Na)+ 457.1340. Found 457.1391.
6-[6-Cyclobutyl-1-(2,3-difluorophenyl)-2-oxo-1,2-dihydropyridin-4-yl] 2-tert-Butoxycarbonyl-5-fluoro-3,4-dihydro-1H-isoquinoline-6-carboxylate (21 m)Compound 21 m was prepared from 20 m according to the procedure for the synthesis of 21h. Yield was 63%. 1H-NMR (CDCl3) δ: 1.50 (9H, s), 1.65–1.88 (4H, m), 2.00–2.14 (2H, m), 2.85–2.95 (2H, m), 3.08–3.22 (1H, m), 3.65–3.75 (2H, m), 4.61–4.70 (2H, m), 6.15–6.21 (1H, m), 6.44–6.50 (1H, m), 6.97–7.10 (2H, m), 7.18–7.37 (2H, m), 7.82–7.94 (1H, m).
tert-Butyl 10-Cyclobutyl-9-(2,3-difluorophenyl)-7,8-dioxo-1,2,4,7,8,9-hexahydro-12-oxa-3,9-diazabenzo[a]anthracene-3-carboxylate (22 m)Compound 22 m was prepared from 21 m according to the procedure for the synthesis of 17a. Yield was 18%. 1H-NMR (CDCl3) δ: 1.52 (9H, s), 1.70–1.95 (4H, m), 2.08–2.22 (2H, m), 3.02–3.11 (2H, m), 3.15–3.28 (1H, m), 3.70–3.84 (2H, m), 4.65–4.75 (2H, m), 6.31 (1H, s), 6.95–7.38 (4H, m), 8.13 (1H, d, J = 8.1 Hz).
10-Cyclobutyl-9-(2,3-difluorophenyl)-1,2,3,4-tetrahydro-9H-12-oxa-3,9-diazabenzo[a]anthracene-7,8-dione (30)Compound 30 was prepared from 22 m according to the procedure for the synthesis of 24. Yield was 69%. mp: 228–232 °C; 1H-NMR (DMSO-d6) δ: 1.50–1.94 (4H, m), 1.96–2.10 (1H, m), 2.17–2.30 (1H, m), 2.90–3.30 (5H, m), 4.01–4.10 (2H, m), 6.56 (1H, s), 7.20 (1H, d, J = 8.1 Hz), 7.32–7.48 (2H, m), 7.60–7.72 (1H, m), 7.83 (1H, d, J = 8.1 Hz); 13C-NMR (DMSO-d6) δ: 16.8 (s), 22.5 (s), 26.8 (s), 26.9 (s), 37.3 (s), 42.1 (s), 47.8 (s), 96.2 (s), 106.6 (s), 118.6 (d), 121.2 (s), 121.9 (s), 123.4 (s), 124.3 (s), 125.0 (dd), 126.2 (d), 126.6 (d), 143.9 (s), 146.0 (dd), 149.9 (dd), 152.1 (s), 158.5 (s), 158.6 (s), 166.3 (s), 172.8 (s); IR (ATR) cm−1: 1682; HR-MS (ESI-TOF) Calcd for C25H20F2N2NaO3 (M + Na)+ 457.1340. Found 457.1388; Anal. Calcd for C25H20F2N2O3: C, 69.12; H, 4.64; N, 6.45. Found: C, 68.92; H, 4.68; N, 6.41; HPLC purity 99.4% (eluent: 10 mM NaH2PO4-Na2HPO4 (pH 7.0)/MeCN = 35/65).
Evaluation of IDUA Activity and Cellular GAG Levels in Hurler Patient-Derived Fibroblasts-
1.
Cell culture
-
Healthy human neonatal dermal fibroblasts (healthy donor fibroblasts) were purchased from Zen-Bio, Inc. (Research Triangle Park, NC, U.S.A.). Hurler patient-derived fibroblasts (GM00798; IDUA-W402X) were obtained from the Coriell Institute for Medical Research (Camden, NJ, U.S.A.). The Coriell Institute for Medical Research guarantees that skin samples were collected under IRB approval and with patient informed consent. The use of human samples was approved by the ethical committee of Kyoto Pharmaceutical Industries, Ltd.
-
2.
Treatment
-
Fibroblasts in MEM (Thermo Fisher Scientific, Waltham, MA, U.S.A.) supplemented with 10% FBS (MEM/10% FBS) or in FibroLife S2 Comp kit medium (LifeLine Cell Technology, Frederick, MD, U.S.A.) were seeded at 1.25 × 105 cells/well (500 µL/well) in 24-well plates and incubated under 5% CO2 at 37 °C for 24 h. Five hundred microliters of the compound dissolved in medium was added to cells, followed by an incubation under 5% CO2 at 37 °C. Cells were treated with the compound for 2 or 6 d.
-
3.
IDUA activity
-
Cells were lysed in 100 µL of Mammalian Cell PE LB (G-Bioscience, St. Louis, MO, U.S.A.). Protein concentrations were measured using the Protein Assay Bicinchoninate Kit (Nacalai Tesque). Twenty microliters of the cell lysate was mixed with 20 µL of 50 µM 4-methylumbelliferyl-α-Iduronide substrate (Glycosynth, Cheshire, U.K.) in 0.1 M sodium formate buffer (pH 3.0) in black half-area 96-well plates (Greiner, Frickenhausen, Germany). The mixture was incubated at 37 °C for 2 h. The reaction was stopped by 50 µL of 0.2 M sodium glycine buffer (pH 10.5). Before and after the incubation, the fluorescence of 4-methylumbelliferone produced by IDUA (Ex 360 nm, Em 460 nm) was measured using a microplate reader (Power Scan HT, BioTek, Winooski, VT, U.S.A.).
-
4.
GAG levels
-
Cells were washed with D-phosphate buffered saline (PBS) and then sonicated and digested by 125 µg/mL papain in 0.2 M sodium phosphate buffer (pH 6.4) at 65 °C for 24 h. After digestion, cell lysates were centrifuged at 10000 × g at 4 °C for 10 min and GAG levels in the supernatants were quantified using the Blyscan Glycosaminoglycan Assay (Biocolor, County Antrim, U.K.). Briefly, cell lysates and Blyscan dye reagent were mixed for 30 min, followed by centrifugation at 12000 rpm for 10 min to precipitate the GAG-dye complex. The precipitate obtained was resuspended in Blyscan dye dissociation reagent. Absorbance at 656 nm was measured using a microplate reader. DNA contents were measured using the DNA Quantity Kit (Cosmo Bio, Tokyo, Japan). Briefly, 40 µL of the lysate, 160 µL of dilution buffer, and 10 µL of color development reagent were mixed in black half-area 96-well plates and fluorescence (Ex. 360 nm, Em. 460 nm) was measured using a microplate reader (Power Scan HT; BioTek, Winooski, VT, U.S.A.).
-
1.
Animals
-
Female IDUA-W392X (TGG→TAG) knock-in mice (C57BL/6J background) were established using the CRISPR/Cas9 system in the Laboratory Animal Resource Center, the University of Tsukuba, and used at 5–7 weeks old. All procedures were conducted in accordance with “Regulation/Procedure on Animal Experimentation at Kyoto Pharmaceutical Industries, Ltd.” and approved by the animal ethical committee of Kyoto Pharmaceutical Industries, Ltd.
-
2.
Administration of KY-640
-
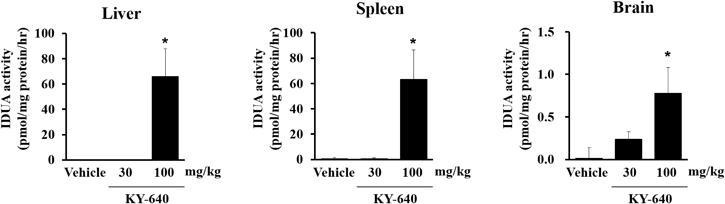
KY-640 (30 and 100 mg/10 mL/kg) was suspended in 0.5% methyl cellulose #400 and orally administered twice a day (BID) for 8 d. During the administration period, body weight and food consumption were monitored and clinical signs were observed daily.
-
3.
IDUA activity
-
Three hours after the last administration on day 8 in the morning, mice were euthanized by exsanguination from the heart under deep anesthesia by the intraperitoneal administration of ketamine (37.5 mg/kg) and xylazine (7.5 mg/kg).
-
Livers, spleens, brains, and kidneys were excised and their wet weights were measured. Relative organ weights were calculated as percentages of body weight [(organ weight/body weight) × 100]. Tissue samples were homogenized in 0.01 M phosphate buffer (pH 7.4) containing 0.1% Triton X-100 (MP Biomedicals, Santa Ana, CA, U.S.A.) and Protease Inhibitor Cocktail (100 mg tissue/mL) (Nacalai Tesque), followed by centrifugation at 20000 × g at 4 °C for 20 min. Protein concentrations in the supernatants were measured using Protein Assay Bicinchoninate Kit. Forty microliters of diluted tissue lysates (100-fold for the liver, 30-fold for the spleen, and 10-fold for the brain) were mixed with 40 µL of 200 µM 4-methylumbelliferyl-α-iduronide substrate in 0.1 M sodium formate buffer (pH 3.5) in 96-well plates. The mixture was incubated at 37 °C for 2, 6, and 24 h. The reaction was stopped by 100 µL of 0.5 M sodium glycine buffer (pH 10.5). The fluorescence of 4-methylumbelliferone produced by IDUA (Ex. 360 nm, Em. 460 nm) was measured using a microplate reader.