Abstract
In the development of anti-severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) drugs, its main protease (Mpro), which is an essential enzyme for viral replication, is a promising target. To date, the Mpro inhibitors, nirmatrelvir and ensitrelvir, have been clinically developed by Pfizer Inc. and Shionogi & Co., Ltd., respectively, as orally administrable drugs to treat coronavirus disease of 2019 (COVID-19). We have also developed several potent inhibitors of SARS-CoV-2 Mpro that include compounds 4, 5, TKB245 (6), and TKB248 (7), which possesses a 4-fluorobenzothiazole ketone moiety as a reactive warhead. In compounds 5 and TKB248 (7) we have also found that replacement of the P1-P2 amide of compounds 4 and TKB245 (6) with the corresponding thioamide improved their pharmacokinetics (PK) profile in mice. Here, we report the design, synthesis and evaluation of SARS-CoV-2 Mpro inhibitors with replacement of a digestible amide bond by surrogates (9–11, 33, and 34) and introduction of fluorine atoms in a metabolically reactive methyl group on the indole moiety (8). As the results, these compounds showed comparable or less potency compared to the corresponding parent compounds, YH-53/5h (2) and 4. These results should provide useful information for further development of Mpro inhibitors.
Introduction
More than three years have passed since the beginning of the pandemic driven by the coronavirus disease of 2019 (COVID-19).1) The infection of a positive-sense single-stranded RNA virus, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is responsible for COVID-19.2,3) At present, there are several authorized drugs for the treatment of COVID-19. These include remdesivir, a repositioned drug that was developed for the treatment of Ebola hemorrhagic fever as an inhibitor against the RNA-dependent RNA polymerase (RdRp)4,5); the SARS-CoV-2 RdRp inhibitor, molnupiravir (LAGEVRIO™, Merck & Co., NJ, U.S.A.)6–8); the SARS-CoV-2 main protease (Mpro) inhibitor nirmatrelvir (1, Paxlovid™, which is a combination of nirmatrelvir and ritonavir tablets, Pfizer Inc., NY, U.S.A., Fig. 1)9) as well as ensitrelvir fumaric acid (Xocova®, Shionogi & Co., Ltd., Osaka, Japan).10) Since SARS-CoV-2 Mpro is an essential enzyme for viral replication and highly conserved among SARS-CoV-2 mutants from an alpha strain to omicron strains,11) this enzyme is an attractive target for drug discovery. Researchers world-wide have put significant effort into the development of a variety of SARS-CoV-2 Mpro inhibitors. These include peptidomimetics, non-peptidic compounds, natural compounds, fullerene derivatives and cyclic peptides.12–28) However, the first generation inhibitor nirmatrelvir (1) can produce drug-resistant SARS-CoV-2 variants.29–32) There are also limitations in combinational dosage of Paxlovid™ with several drugs due to its ritonavir-boosting, which causes inhibition of CYP3A4 that can result in undesirable blood concentration enhancements of other drugs.33) Therefore, development of biostable SARS-CoV-2 Mpro inhibitors is still urgently needed to increase available drug repertoires. In addressing this challenge, we have been developing reversible covalent Mpro inhibitors based on YH-53/5h (2), which was previously developed by Hayashi et al. as a SARS-CoV Mpro inhibitor34–36) (Fig. 1). Our efforts resulted in the highly potent SARS-CoV-2 Mpro inhibitors 4 and 5, and subsequently to the more promising Mpro inhibitors TKB245 (6) and TKB248 (7).37–39) These compounds all possess a 4-fluorobenzothiazolyl ketone group as a reactive warhead that includes a fluorine atom at the 4-position of the benzothiazole moiety, which is a key for improved potency.37) In our current study, we design and synthesize biostable Mpro inhibitors that incorporate amide bond surrogates and introduce fluorine atoms.
Results and Discussion
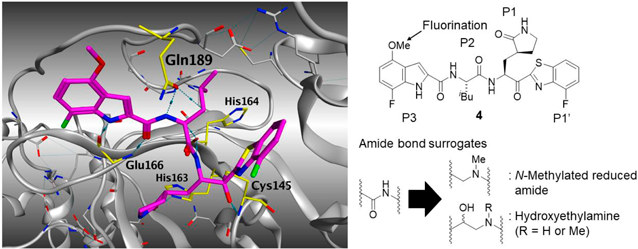
Concept of Compound DesignIn our previous study, we have found that replacement of the P1-P2 amide bond in YH-53/5h (2), 4, and TKB245 (6) with the corresponding thioamide structure to yield compounds 3, 5, and TKB248 (7), improved their in vivo pharmacokinetics (PK) and increased cellular concentrations.37,38) This suggested that modification of the biocleavable amide bonds between P1–P2 sites could potentially afford more drug-like compounds with preferable PK relative to the amide-type compounds. It has been reported that the methoxy group in the indole ring of YH-53/5h (2) can be metabolized in cryopreserved human and rat hepatocytes.36) Replacement of this methoxy group with a trifluoromethoxy group (2-CF3) prolonged its half-life in mice, and the potency of 2-CF3 was comparable to that of the parent YH-53/5h (2).37) Therefore, we synthesized the difluoromethoxy indole derivative 8 with a 4-fluorobenzothiazolyl ketone moiety (Chart 1) to improve potency and biostability (Fig. 2, right). The co-crystal structure of SARS-CoV-2 Mpro with compound 4 shows that 4 forms multiple hydrogen bonds within the catalytic pocket of Mpro and a covalent bond with Cys145 (Fig. 2, left). Therefore, we introduced an N-methylated reduced amide unit and a hydroxyethyl amine unit as amide bond surrogates between the P1-P2 sites (Fig. 2, right) to obtain compounds 9, 10 and 11 (Charts 2–4 and Figs. 3 and 4). Introduction of an N-methylated reduced amide moiety can retain the number of carbons in backbone structure, while that of a hydroxylethylamino unit contains results in one additional carbon atom. The hydroxylethylamine unit is a well-known transition state mimetic in human immunodeficiency virus type 1 (HIV-1) aspartyl protease inhibitors, such as Darunavir.40–49) Therefore, this motif would be worth introducing into SARS-CoV-2 Mpro inhibitors as a non-hydrolysable amide bond surrogate that circumvents biodegradation of the P1-P2 amide bonds.

Where indicated, compound purities were determined to be >95% by analytical reverse phase (RP)-HPLC or NMR analysis. Experimental procedures of the synthesis of all of compounds, including characterization data are provided in the Supplementary Materials. The syntheses of the representative compounds 8, 9, 10, and 11 are shown in Charts 1–4. In the synthesis of compound 8, L-Leu-OtBu hydrochloride (12) was coupled with 4-(difluoromethoxy)-1H-indole-2-carboxylic acid (13) using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI·HCl) in the presence of 1-hydroxybenzotriazole monohydrate (HOBt·H2O) and N,N-diisopropylethylamine (DIPEA) to provide the tBu ester 14. The carboxylic acid 15 was prepared by treatment of the tBu ester 14 with hydrochloric acid and coupled with amine 1737) using 1-[(1-(cyano-2-ethoxy-2-oxoethylideneamino-oxy)-dimethylaminomorpholino)]uronium hexafluorophosphate (COMU) in the presence of DIPEA to afford the target compound 8 (Chart 1).
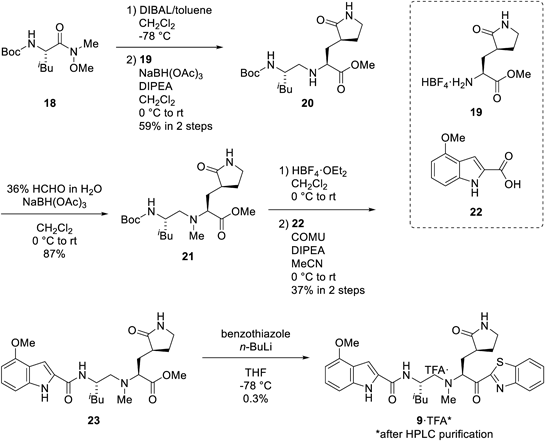
Reduction of the Weinreb amide 18 by treatment with diisobutylaluminium hydride (DIBAL) and subsequent reductive amination with amine 1937) provided amine 20. The second reductive amination of the resulting 20 with formaldehyde, deprotection of tert-butoxycarbonyl (Boc) group by HBF4·OEt2 and coupling with indole 22 using COMU in the presence of DIPEA gave the methyl ester 23. Treatment with lithiated benzothiazole afforded the target compound 9 (Chart 2).
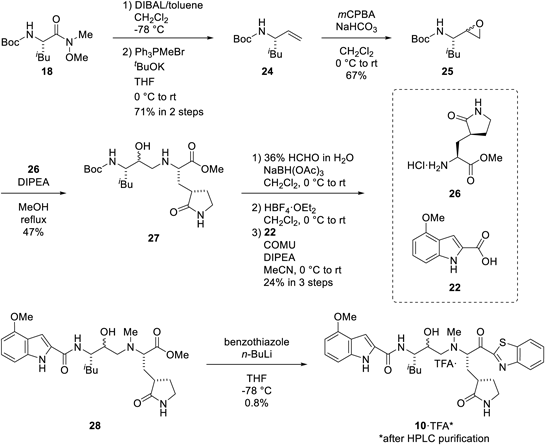
Reduction of the Weinreb amide 18 by treatment of DIBAL and subsequent Wittig Reaction with triphenylmethylphosphonium bromide provided alkene 24. Epoxidation of alkene 24 using m-chloroperoxybenzoic acid (mCPBA) followed by nucleophilic ring opening of the resulting epoxide 25 using amine 26 gave the hydroxyethylamine-contained compound 27. The amino group of 27 was then methylated by reductive amination with formaldehyde followed by Boc deprotection with HBF4·OEt2. Coupling with indole 22 using COMU in the presence of DIPEA provided the methyl ester 28. Treatment of ester 28 with lithiated benzothiazole afforded the target compound 10 (Chart 3).
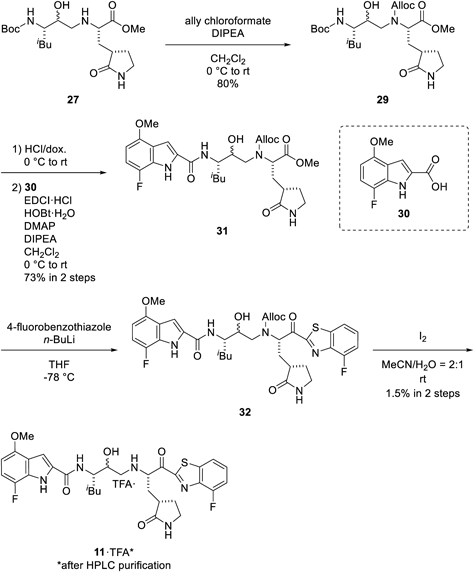
The amino group of amine 27 was protected using allyl chloroformate to give the allyloxycarbonyl (Alloc)-protected hydroxyethylamine 29. The Boc group of amine 29 was deprotected with hydrochloric acid, and the resulting amine was then coupled with indole 30 using EDCI·HCl in the presence of HOBt·H2O, 4-dimethylaminopyridine (DMAP), and DIPEA to provide the methyl ester 31. Treatment of ester 31 with lithiated 4-fluorobenzothiazole obtained ketone 32. Deprotection of the Alloc group of the ketone 32 by treatment with iodine under aqueous conditions50) afforded the target 11 (Chart 4).
All the final compounds, 8, 9, 10, 11, 33, and 34, were purified by silica gel column chromatography followed by preparative RP-HPLC and semi-preparative chiral normal phase (NP)-HPLC purifications.
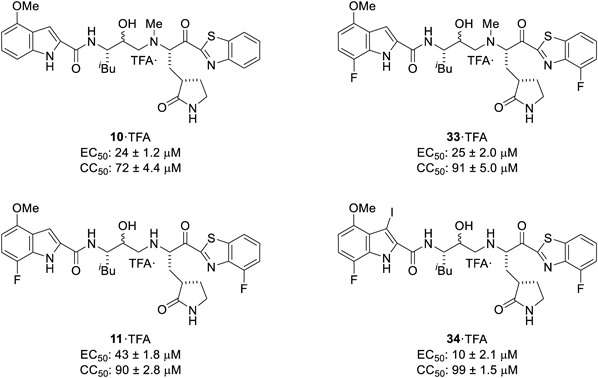
Structure–Activity Relationship StudiesPrevious work has shown that a benzothiazolyl ketone moiety at the P1′ site of SARS-CoV-2 Mpro inhibitors functions as an ideal warhead that can reversibly form a covalent bond with the catalytic Cys145 residue of Mpro and occupy a larger volume in the catalytic pocket than the cyano group in nirmatrelvir (1).12,37,38) Accordingly, in our current study benzothiazolyl and 4-fluorobenzothyazolyl ketones were used as warhead structures, while N-methylated reduced amide and hydroxyethylamine units were introduced as amide bond surrogates between the P1-P2 sites. The EC50 and 50% cytotoxic concentration (CC50) values of the new compounds were determined with quantitative RT-PCR (RT-qPCR) and WST-8 assays, respectively, using VeroE6 cells (see the Experimental section).12) The difluoromethoxy indole-containing compound 8 showed comparable potency with that of YH-53/5h (2), whereas the N-methylated reduced amide-type derivative 9 exhibited dramatically decreased potency (Fig. 3). This suggested that a hydrogen bond between the hydrogen atom of the α-amino group at the P1 site of Mpro inhibitors and the His164 is essential for potent anti-SARS-CoV-2 activities. Although we attempted to synthesize the N-free reduced amide-type Mpro inhibitors, these compounds appeared to be chemically unstable and we were not able to isolate them in sufficient purity. Therefore, we concluded that N-free reduced amide-type Mpro inhibitors are not viable drug candidates.
The N-methylated hydroxyethylamine type derivatives 10 and 33 and hydroxyethylamine type derivatives 11 and 34 were investigated next (Fig. 4). In contrast to the pattern shown by YH-53/5h (2) and compound 4, compound 33, which represents a derivative of compound 10 having fluorine substitutents at the indole 7-position and the 4-position of the benzothiazole moiety, failed to show improved potency relative to the parent compound 10. Comparison with compounds 33 and 11 indicates that N-methylation of the α-amino group at the P1 site does not attenuate the potency. The iodinated compound 34, which was obtained as a byproduct during deprotection of the N-Alloc group by iodine treatment, showed higher potency than compound 11. The reasons for this remain unclear. The tested compounds 10, 11, 33 and 34 all showed decreased potency relative to the parent lead compound YH-53/5h (2), while also exhibiting varying degrees of cytotoxicity (CC50 < 100 µM). These results suggest that the hydroxyethylamine unit is not suitable as an amide bond surrogate between P1-P2 sites of SARS-CoV-2 Mpro inhibitors and that further optimization is required to develop more potent and biostable Mpro inhibitors. All of the tested compounds were also subjected to immunostaining experiments to confirm their cytotoxicities (Supplementary Fig. S1).
Conclusion
The COVID-19 pandemic caused by SARS-CoV-2 infection has now been continuing for more than three years. Numerous SARS-CoV-2 vaccines and drugs targeting SARS-CoV-2 enzymes have been developed to date. However, moderate efficacy and the limitations of combinational treatments, provide a compelling rationale for the further development of new anti-COVID-19 drugs. Herein, we present our efforts to develop potent and biostable SARS-CoV-2 Mpro inhibitors by fluorination and introduction of amide bond surrogates at metabolically labile positions. Our approach has been to initially design a difluoromethoxyindole-containing derivative 8 with a 4-fluorobenzothiazolyl ketone as a warhead structure. The intent was to improve the potency and biostability by fluorination. We examined N-methylated reduced amides typified by 9, in which the metabolically unstable amide bond between the P1-P2 positions has been converted into tertiary amines while retaining the same number backbone carbon numbers. These derivatives showed moderate-to-reduced potencies (Fig. 3). This suggested the difluoromethoxyindole moiety is suitable for use in Mpro inhibitors and that the hydrogen bond between the α-amino group at the P1 and the His164 is indispensable. This is consistent with our previous findings that the thioamide derivatives 5 and TKB248 (7) having a hydrogen atom on the α-amino group at the P1 site exhibit comparable potencies with those of these parent compounds 4 and TKB245 (6), respectively.37–39) The hydroxyethylamine unit is recognized as a transition state-mimicking amide bond surrogate that is widely utilized in the development of aspartic protease inhibitors targeting HIV-1 protease40–49) and β-secretase (BACE1; β-site APP cleaving enzyme 1) for the treatment of Alzheimer’s disease.51,52) Therefore, we examined the hydroxyethylamine unit to SARS-CoV-2 inhibitors as an amide bond surrogate by synthesizing the N-methylated hydroxyethylamine-type derivatives 10 and 33 and the hydroxyethylamine-type derivatives 11 and 34. We found that these hydroxyethylamine-type derivatives showed attenuated potencies relative to the parent compounds YH-53/5h (2) and 4. This suggests that the hydroxyethylamine unit is not suitable as an amide bond surrogate between the P1-P2 sites. Our data should facilitate the design of further Mpro inhibitors as drug candidates for COVID-19 therapy. Further research in this direction is ongoing in our laboratory.
Experimental
Compound SynthesisThe synthetic methods for representative compounds 8, 9, 10, and 11 are described in Charts 1–4. The purity of all the final compounds was measured by analytical RP-HPLC or NMR is >95%. Experimental procedures including characterization data, NMR spectra and analytical RP-HPLC charts of the purified compounds are provided in the Supplementary Materials.
Cells, Viruses and Test CompoundsVeroE6 cells were obtained from the American Type Culture Collection (ATCC) (CRL-1586) (Manassas, VA, U.S.A.) and were maintained in Dulbecco’s modified Eagle’s medium (D-MEM) supplemented with 10% fetal bovine serum (FBS), 100 µg/mL of penicillin, and 100 µg/mL of streptomycin. The SARS-CoV-2 strain JPN/TY/WK-521 (SARS-CoV-2WK-521, ancestral Wuhan strain, GISAID Accession ID; EPI_ISL_408667) was obtained from the National Institute of Infectious Diseases (Tokyo, Japan). Each compound was dissolved in dimethyl sulfoxide (DMSO) at 20 mM as a stock solution.
Anti-SARS-CoV-2 and Cytotoxicity AssaysFor antiviral assays, cells were seeded in a 96-well plate (2 × 104 cells/well) and incubated. After 1 d, virus was inoculated into cells at each multiplicity of infection (MOI): SARS-CoV-2WK-521 (0.33). After an additional 3 d, cell culture supernatants were harvested and viral RNA was extracted using a QIAamp viral RNA minikit (Qiagen, Hilden, Germany), and RT-qPCR was then performed using One Step PrimeScript III RT-qPCR mix (TaKaRa Bio, Shiga, Japan) following the instructions of the manufacturers. The primers and probe used for detecting SARS-CoV-2 nucleocapsid53) were 5′-AAATTTTGGGGACCAGGAAC-3′ (forward), 5′-TGGCAGCTGTGTAGGTCAAC-3′ (reverse), and 5′-FAM-ATGTCGCGCATTGGCATGGA-black hole quencher 1 (BHQ1)-3′ (probe). To determine the cytotoxicity of each compound, cells were seeded in a 96-well plate (2 × 104 cells/well). One day later, various concentrations of each compound were added, and cells were then incubated for an additional 3 d. Values of the CC50 were determined based on the WST-8 assays using Cell Counting Kit-8 (Dojindo, Kumamoto, Japan).
Acknowledgments
This work was supported in part by Research Projects on COVID-19, Japan Agency for Medical Research and Development (AMED) 20fk0108510 and 21fk0108480 (H.M., H.T.); JSPS KAKENHI Grant Numbers 20H03362 (H.T.), 22K15243 (K.T.), and 23K14318 (T.K.); AMED under Grant Number JP23ama121043 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS) (H.T.); JPMJFS2109 (MEXT, the establishment of university fellowships towards the creation of science technology innovation) (T.I., Y.M.). This research is based on the Cooperative Research Project of Research Center for Biomedical Engineering. The authors thank Dr. Terrence R. Burke, Jr., NCI/NIH, for editing this manuscript.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials. Supplementary Materials in PDF format contains synthetic experimental procedures, compound characterization (1H-NMR, 13C-NMR, and HRMS) and immunostaining experimental data (Supplementary Fig. S1).
References
- 1) World Health Organization, “WHO Coronavirus (COVID-19) Dashboard.”: ‹https://covid19.who.int/›, cited 28 July, 2023.
- 2) Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., N. Engl. J. Med., 382, 727–733 (2020).
- 3) Mitsuya H., Kokudo N., Glob. Health Med., 2, 53–55 (2020).
- 4) Pardo J., Shukla A. M., Chamarthi G., Gupte A., Drugs Context, 9, 2020 (2020).
- 5) U.S. Food & Drug Administration. “FDA approves first treatment for COVID-19.”: ‹https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19›, cited 28 July, 2023.
- 6) Merck & Co, Inc., “Merck and Ridgeback’s Investigational Oral Antiviral Molnupiravir Reduced the Risk of Hospitalization or Death by Approximately 50% Compared to Placebo for Patients with Mild or Moderate COVID-19 in Positive Interim Analysis of Phase 3 Study.”: ‹https://www.merck.com/news/merck-and-ridgebacks-investigational-oral-antiviral-molnupiravir-reduced-the-risk-of-hospitalization-or-death-by-approximately-50-percent-compared-to-placebo-for-patients-with-mild-or-moderat/›, cited 28 July, 2023.
- 7) World Health Organization, “WHO updates its treatment guidelines to include molnupiravir.”: ‹https://www.who.int/news/item/03-03-2022-molnupiravir›, cited 28 July, 2023.
- 8) McIntosh J. A., Benkovics T., Silverman S. M., et al., ACS Cent. Sci., 7, 1980–1985 (2021).
- 9) Owen D. R., Allerton C. M. N., Anderson A. S., et al., Science, 374, 1586–1593 (2021).
- 10) Unoh Y., Uehara S., Nakahara K., et al., J. Med. Chem., 65, 6499–6512 (2022).
- 11) Lee J. T., Yang Q., Gribenko A., Perrin B. S. Jr., Zhu Y., Cardin R., Liberator P. A., Anderson A. S., Hao L., MBio, 13, e0086922 (2022).
- 12) Hattori S., Higashi-Kuwata N., Hayashi H., et al., Nat. Commun., 12, 668 (2021).
- 13) Hoffman R. L., Kania R. S., Brothers M. A., et al., J. Med. Chem., 63, 12725–12747 (2020).
- 14) Qiao J., Li Y.-S., Zeng R., et al., Science, 371, 1374–1378 (2021).
- 15) Bai B., Belovodskiy A., Hena M., et al., J. Med. Chem., 65, 2905–2925 (2022).
- 16) Xia Z., Sacco M., Hu Y., Ma C., Meng X., Zhang F., Szeto T., Xiang Y., Chen Y., Wang J., ACS Pharmacol. Transl. Sci., 4, 1408–1421 (2021).
- 17) Ma C., Xia Z., Sacco M. D., Hu Y., Townsend J. A., Meng X., Choza J., Tan H., Jang J., Gongora M. V., Zhang X., Zhang F., Xiang Y., Marty M. T., Chen Y., Wang J., J. Am. Chem. Soc., 143, 20697–20709 (2021).
- 18) Wang H., Pei R., Li X., Deng W., Xing S., Zhang Y., Zhang C., He S., Sun H., Xiao S., Xiong J., Zhang Y., Chen X., Wang Y., Guo Y., Zhang B., Shang L., Eur. J. Med. Chem., 238, 114458 (2022).
- 19) Ma Y., Yang K. S., Geng Z. Z., Alugubelli Y. R., Shaabani N., Vatansever E. C., Ma X. R., Cho C.-C., Khatua K., Xiao J., Blankenship L. R., Yu G., Sankaran B., Li P., Allen R., Ji H., Xu S., Liu W. R., Eur. J. Med. Chem., 240, 114570 (2022).
- 20) Vankadara S., Dawson M. D., Fong J. Y., Oh Q. Y., Ang Q. A., Liu B., Chang H. Y., Koh J., Koh X., Tan Q. W., Joy J., Chia C. S. B., ACS Med. Chem. Lett., 13, 1345–1350 (2022).
- 21) Hirose Y., Shindo N., Mori M., et al., J. Med. Chem., 65, 13852–13865 (2022).
- 22) Panarese J. D., Davis D., Kenton N. T., Bartlett S., Rafferty S., Or Y. S., WO2022020242 (2021).
- 23) Issa S. S., Sokornova S. V., Zhidkin R. R., Matveeva T. V., Plants, 11, 1862 (2022).
- 24) Katagishi D., Yasuda D., Takahashi K., Nakamura S., Mashino T., Ohe T., Bioorg. Med. Chem. Lett., 80, 129121 (2023).
- 25) Miura T., Malla T. R., Owen C. D., Tumber A., Brewitz L., McDonough M. A., Salah E., Terasaka N., Katoh T., Lukacik P., Strain-Damerell C., Mikolajek H., Walsh M. A., Kawamura A., Schofield C. J., Suga H., Nat. Chem., 15, 998–1005 (2023).
- 26) Maehara S., Kobayashi M., Kuwada M., Choshi T., Inoue H., Hieda Y., Nishiyama T., Hata T., Chem. Pharm. Bull., 70, 195–198 (2022).
- 27) Hasegawa T., Imamura R. M., Suzuki T., Hashiguchi T., Nomura T., Otsuguro S., Maenaka K., Sasaki M., Orba Y., Sawa H., Sato A., Okabe T., Nagano T., Kojima H., Chem. Pharm. Bull., 70, 199–201 (2022).
- 28) Miwa K., Guo Y., Hata M., Yamamoto N., Hoshino T., Chem. Pharm. Bull., 71, 360–367 (2023).
- 29) Iketani S., Mohri H., Culbertson B., Hong S. J., Duan Y., Luck M. I., Annavajhala M. K., Guo Y., Sheng Z., Uhlemann A. C., Goff S. P., Sabo Y., Yang H., Chavez A., Ho D. D., Nature (London), 613, 558–564 (2023).
- 30) Ullrich S., Ekanayake K. B., Otting G., Nitsche C., Bioorg. Med. Chem. Lett., 62, 128629 (2022).
- 31) Hu Y., Lewandowski E. M., Tan H., Zhang X., Morgan R. T., Zhang X., Jacobs L. M. C., Butler S. G., Gongora M. V., Choy J., Deng X., Chen Y., Wang J., bioRχiv., DOI: 10.1101/2022.06.28.497978 (2022), cited 29 July, 2023.
- 32) Yang K. S., Leeuwon S. Z., Xu S., Liu W. R., J. Med. Chem., 65, 8686–8698 (2022).
- 33) National Institutes of Health, “Ritonavir-Boosted Nirmatrelvir (Paxlovid).”: ‹https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/ritonavir-boosted-nirmatrelvir--paxlovid-/›, cited 29 July, 2023.
- 34) Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Chen S. E., Naser-Tavakolian A., Schön A., Freire E., Hayashi Y., Eur. J. Med. Chem., 68, 372–384 (2013).
- 35) Konno S., Thanigaimalai P., Yamamoto T., Nakada K., Kakiuchi R., Takayama K., Yamazaki Y., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S.-E., Freire E., Hayashi Y., Bioorg. Med. Chem., 21, 412–424 (2013).
- 36) Konno S., Kobayashi K., Senda M., et al., J. Med. Chem., 65, 2926–2939 (2022).
- 37) Tsuji K., Ishii T., Kobayakawa T., et al., iScience, 25, 105365 (2022).
- 38) Higashi-Kuwata N., Tsuji K., Hayashi H., et al., Nat. Commun., 14, 1076 (2023).
- 39) Tsuji K., Ishii T., Kobayakawa T., Higashi-Kuwata N., Shinohara K., Azuma C., Miura Y., Nakano H., Wada N., Hattori S., Bulut H., Mitsuya H., Tamamura H., J. Med. Chem., 66, 13516–13529 (2023).
- 40) Roberts N. A., Martin J. A., Kinchington D., Broadhurst A. V., Craig J. C., Duncan I. B., Galpin S. A., Handa B. K., Kay J., Kröhn A., Lambert R. W., Merrett J. H., Mills J. S., Parkes K. E. B., Redshaw S., Ritchie A. J., Taylor D. L., Thomas G. J., Machin P. J., Science, 248, 358–361 (1990).
- 41) Ghosh A. K., Thompson W. J., Holloway M. K., McKee S. P., Duong T. T., Lee H. Y., Munson P. M., Smith A. M., Wai J. M., Darke P. L., Zugay J. A., Emini E. A., Schleif W. A., Huff J. R., Anderson P. S., J. Med. Chem., 36, 2300–2310 (1993).
- 42) Dorsey B. D., Levin R. B., McDaniel S. L., Vacca J. P., Guare J. P., Darke P. L., Zugay J. A., Emini E. A., Schleif W. A., Quintero J. C., Lin J. H., Chen I.-W., Holloway M. K., Fitzgerald P. M. D., Axel M. G., Ostovic D., Anderson P. S., Huff J. R., J. Med. Chem., 37, 3443–3451 (1994).
- 43) Kim E. E., Baker C. T., Dwyer M. D., Murcko M. A., Rao B. G., Tung R. D., Navia M. A., J. Am. Chem. Soc., 117, 1181–1182 (1995).
- 44) Kaldor S. W., Kalish V. J., Davies J. F. II, Shetty B. V., Fritz J. E., Appelt K., Burgess J. A., Campanale K. M., Chirgadze N. Y., Clawson D. K., Dressman B. A., Hatch S. D., Khalil D. A., Kosa M. B., Lubbehusen P. P., Muesing M. A., Patick A. K., Reich S. H., Su K. S., Tatlock J. H., J. Med. Chem., 40, 3979–3985 (1997).
- 45) Tamamura H., Koh Y., Ueda S., Sasaki Y., Yamasaki T., Aoki M., Maeda K., Watai Y., Arikuni H., Otaka A., Mitsuya H., Fujii N., J. Med. Chem., 46, 1764–1768 (2003).
- 46) Koh Y., Nakata H., Maeda K., Ogata H., Bilcer G., Devasamudram T., Kincaid J. F., Boross P., Wang Y.-F., Tie Y., Volarath P., Gaddis L., Harrison R. W., Weber I. T., Ghosh A. K., Mitsuya H., Antimicrob. Agents Chemother., 47, 3123–3129 (2003).
- 47) Ghosh A. K., Ramu Sridhar P., Kumaragurubaran N., Koh Y., Weber I. T., Mitsuya H., ChemMedChem, 1, 939–950 (2006).
- 48) Ghosh A. K., Sridhar P. R., Leshchenko S., Hussain A. K., Li J., Kovalevsky A. Y., Walters D. E., Wedekind J. E., Grum-Tokars V., Das D., Koh Y., Maeda K., Gatanaga H., Weber I. T., Mitsuya H., J. Med. Chem., 49, 5252–5261 (2006).
- 49) Ghosh A. K., Dawson Z. L., Mitsuya H., Bioorg. Med. Chem., 15, 7576–7580 (2007).
- 50) Szumigala R. H. Jr., Onofiok E., Karady S., Armstrong J. D. III, Miller R. A., Tetrahedron Lett., 46, 4403–4405 (2005).
- 51) Kobayashi K., Otani T., Ijiri S., Kawasaki Y., Matsubara H., Miyagi T., Kitajima T., Iseki R., Ishizawa K., Shindo N., Okawa K., Ueda K., Ando S., Kawakita M., Hattori Y., Akaji K., Bioorg. Med. Chem., 50, 116459 (2021).
- 52) Otani T., Hattori Y., Akaji K., Kobayashi K., Bioorg. Med. Chem., 52, 116517 (2021).
- 53) Shirato K., Nao N., Katano H., Takayama I., Saito S., Kato F., Katoh H., Sakata M., Nakatsu Y., Mori Y., Kageyama T., Matsuyama S., Takeda M., Jpn. J. Infect. Dis., 73, 304–307 (2020).